
阿尔茨海默病信号通路研究进展及对策研究
2021-02-07 06:28谢林江李劲松
中国药理学通报 2021年2期
谢林江,邓 婷,徐 颖,李劲松,孙 涛
(1. 成都中医药大学 中药材标准化教育部重点实验室,西南特色中药资源国家重点实验室,四川 成都 611137;2. 广元市第一人民医院,四川 广元 628017)
阿尔茨海默病(Alzheimer’s disease,AD) 是一种神经退行性疾病,其特征为记忆力及其它认知功能渐进性丧失。如今已经明确阐明AD的两个核心病理表现为淀粉样斑块(amyloid plaques)和神经元缠结(neurofibrillary tangles)的积累。近年来,基于对细胞信号转导通路研究的不断深入,许多关于AD的致病机制被逐步揭示,其中影响AD的信号通路主要有PI3K/Akt、Wnt、Notch、MAPK、NF-κB等。这些细胞信号转导通路中相关元件有可能成为潜在治疗路径及靶点。
1 AD相关PI3K/Akt信号转导通路
1.1 组成和调控磷脂酰肌醇-3-激酶(phosphatidylinositol 3-kinase,PI3K)是一组与质膜相关的脂质激酶,其组成为p110催化亚基、p55调节亚基和p85调节亚基。PI3K的募集由酪氨酸激酶受体(receptor tyrosine kinases,RTKs)激活引起,进而PI3K会催化磷酯酰肌醇二磷酸(phosphatidylinositol 4,5-bisphosphat,PIP2)生成磷脂酰肌醇-3,4,5-三磷酸(phosphatidylinositol-(3,4,5)-trisphosphate,PIP3),PIP3是一种第二信使,可进一步激活其下游蛋白激酶B(protein kinase B,PKB/AKT),进而定位于细胞膜上,激活细胞存活和细胞生长途径,磷酸酶张力蛋白(phosphatase and tensin homolog,PTEN)通过让PIP3去磷酸化,充当PI3K途径的负向调节剂。AKT激酶家族成员有3个,分别为AKT1/PKBα、AKT2/PKBβ和AKT3/PKBγ,AKT中两个残基苏氨酸308(Thr 308)和丝氨酸473(Ser 473)磷酸化后,AKT激酶功能被激活,活化的AKT可调节信号通路中的下游蛋白如雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、糖原合酶激酶3β(glycogen synthase kinase 3β,GSK-3β)、环磷腺苷效应元件结合蛋白(cAMP response element binding protein,CREB)等,其中mTOR与促炎因子释放有关,CREB影响神经元存活、学习和记忆能力,GSK-3β与Tau蛋白过度磷酸化有关[1-2]。研究发现AKT只对神经元的存活有调节作用,对神经突触生长和分化等无调节作用,可能通过直接抑制促凋亡转录因子Forkhead或凋亡蛋白Bad,间接通过使GSK-3β Ser9位点磷酸化抑制其活性,或通过阻断神经元主要凋亡通路JNK-p53-Bax的功能上调抗凋亡因子IAP、Bcl-2或Bcl-xl水平来抑制神经细胞凋亡[2]。选择性PI3K抑制剂LY294002(合成化合物)可逆转花青素的神经保护作用并增加凋亡蛋白caspase-3/-7的表达[3]。
1.2 信号通路与ADAli等[3]发现,花青素通过PI3K/Akt/GSK3β信号通路降低β淀粉样蛋白寡聚体(amyloid beta oligomer,AβO)刺激内源性抗氧化系统的核因子红系2相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)和血红素加氧酶1(heme oxygenase-1,Nrf2/HO-1)诱导的ROS水平升高和氧化应激反应。Xu等[4]发现,由神经炎症诱导激活的A1星形胶质细胞会导致AD中神经元和少突胶质细胞的死亡,乳脂小球表皮生长因子8(milk fat globule epidermal growth factor 8,MFG-E8)在小胶质细胞条件培养基(microglia conditioned medium,MCM)刺激下可以通过上调PI3K/Akt通路抑制A1星形细胞活化,可能是治疗AD神经炎症的潜在药物治疗靶标。Cui等[5]发现,内质网应激与AD神经元丢失有关,而PTEN抑制剂双过氧化双钾-(5-羟基吡啶-2-羧基)-氧杂钒酸盐(bpv)能通过PI3K/Akt途径降低内质网应激相关蛋白的表达减少神经细胞凋亡。Wang等[6]发现,小檗碱能通过激活PI3K/Akt信号通路使Tau蛋白去磷酸化来改善神经元损伤。神经元损伤可导致认知能力、记忆力、学习能力下降和一些情绪变化,成人神经元具有自我修复能力,但随着年龄增长修复能力下降,造成神经元进行性损伤进而引起阿尔茨海默病。上述药物基于激活PI3K/Akt信号通路,降低AβO引起的氧化应激,抑制A1星形细胞活化改善神经炎症导致的神经细胞死亡,抑制内质网应激减少神经细胞凋亡以及使Tau蛋白去磷酸化改善神经元损伤。
2 AD相关Wnt信号转导通路
2.1 组成与调控Wnt蛋白是一种富含半胱氨酸的分泌型糖蛋白家族,通过与细胞表面的几种受体和协同受体结合而激活不同的细胞内信号传导途径。其信号转导途径主要分为以下3种:Wnt/β-catenin途径,平面细胞极性(planar cell polarity,PCP)途径和Wnt/Ca2+途径,这些途径可改变基因表达和使细胞骨架重组。当Wnt配体与其受体卷曲蛋白(frizzled,Fzd)和协同受体低密度脂蛋白受体5/6(low-density lipoprotein receptor 5/6,LRP 5/6)结合后,支架蛋白Disheveled(Dvl)和Axin在细胞膜上募集,同时形成信号小体,进而允许β-catenin在细胞质中积累,进一步发生核易位,并与T细胞特异性转录因子(TCF)/淋巴增强因子(LEF)和其他辅因子结合,从而调节Wnt靶基因的转录。在没有Wnt配体的情况下,在细胞质中酪蛋白激酶 1α(casein kinase-1α,CK-1α)和GSK-3β等激酶能使β-catenin磷酸化而降解。第二个Wnt/PCP途径的激活则不需要协同受体LRP 5/6的参与,该途径首先通过激活小G蛋白家族(small GTPases)中的RhoA和Rac1,进一步激活其下游的Rho相关卷曲螺旋形成蛋白激酶(Rho associated coiledcoil forming protein kinase,ROCK)和c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)。第三个途径是Wnt/Ca2+级联反应,其中Wnt与卷曲蛋白受体的结合使细胞内存储中Ca2+释放以及磷脂酶C(phospholipase C,PLC)、钙调蛋白激酶II(calmodulin-dependent protein kinase II,CaMKII)和蛋白激酶C(protein kinase C,PKC)的活化,进而调节转录和肌动蛋白重塑。Wnt信号通路是中枢神经系统发育的基础,其信号保持良好平衡是维持成熟突触所必需,且Wnt信号转导与树突和突触形成,突触可塑性和维持以及轴突寻路有关[7]。Dkk1是一种衰老小鼠大脑中内源性分泌Wnt拮抗剂,作用于LRP 5/6,减少Wnt蛋白与Fzd和LRP 5/6的结合来抑制Wnt/β-catenin信号转导,进而致使突触损失和促进Aβ蛋白产生[8]。
2.2 信号通路与ADHuang等[9]发现,氟西汀可引起2A型蛋白磷酸酶(protein phosphatase 2A,PP2A)激活,使β-catenin活性水平提高,GSK-3β活性降低,进而激活Wnt/β-catenin信号通路,发挥预防Aβ纤维诱导的神经变性和改善神经元变性和突触损伤的作用。Sellers等[10]发现,Aβ蛋白诱导Dkk1具有双重作用,它不仅抑制经典的Wnt途径,同时激活Wnt-PCP途径,而ROCK抑制剂法舒地尔(Fasudil)能够拮抗Wnt/PCP信号通路,降低Tau蛋白磷酸化水平从而预防Aβ诱导的认知障碍和突触损伤。Zhang等[11]发现,L-3-正丁基苯酞(L-NBP)治疗能显著增加突触数量和厚度,增加突触后密度蛋白95(postsynaptic density protein-95,PSD-95)、突触素(synaptophysin,SYN)以及β-catenin的表达水平,可能与Wnt/β-catenin信号通路激活有关。正确的突触功能是大脑发挥正常功能的必要条件,突触为神经元细胞高度分化至关重要的部分,是神经元细胞之间交流的主要场所。神经退行性疾病均表达突触功能障碍这一病理机制,突触功能障碍将引起神经元结构和功能逐渐丧失最终导致神经元死亡。上述药物基于激活Wnt/β-catenin信号通路增加突触数量、厚度和增加PSD-95、SYN表达水平预防Aβ诱导的神经变性以及改善神经元变性和突触损伤,基于抑制Wnt/PCP信号通路降低Tau蛋白磷酸化水平减少突触损伤。
3 AD相关Notch信号转导通路
3.1 组成与调控Notch信号转导是一种在进化上极其保守的途径,在细胞命运选择、代谢、肿瘤发生和器官发育中具有至关重要的作用。Notch受体是一种跨膜蛋白,其胞外域由大量EGF重复序列和负调控区(negative regulatory region,NRR)组成,哺乳动物拥有4个不同的Notch受体(Notch1-4),Notch受体与邻近细胞上的配体结合后启动Notch信号转导,该配体驱动Notch受体的有序蛋白水解切割和Notch细胞内结构域(Notch intracellular domain,NICD)的最终释放,然后NICD易位至细胞核,在那里募集转录复合物以激活下游靶标基因。Notch信号与神经干细胞(neural stem cells,NSCs)的命运和维持有关,能够调节新生神经元的树突形态,并且通过改善树突可塑性促进记忆形成[12]。基于Notch信号通路暂未发现选择性抑制剂,目前常用抑制剂为DAPT(合成化合物),一种γ-分泌酶抑制剂,而γ-分泌酶潜在底物除Notch蛋白外还有60多种其它蛋白[13]。
3.2 信号通路与ADLi等[13]发现,姜黄素通过激活Notch信号通路,促进NSCs增殖改善认知障碍。Zhang等[14]发现,褪黑素体内外给药能阻止Aβ1-42诱导的Notch1、Notch1跨膜片段(NTMF)、Notch1活性形式(NICD)的表达降低,改善记忆缺陷与神经受损,揭示褪黑素的神经保护作用可能与调节Notch信号通路有关。Hu等[15]发现,黄芪甲苷(ASI)可使Notch-1和NICD的表达增加,通过激活Notch信号通路诱导NSCs增殖和分化,改善学习和记忆能力。NSCs分化为神经元,并整合到神经网络中称为神经发生,是大脑中产生新神经元的过程。新神经元的产生与正常的学习和遗忘过程有关,新神经元通过调节海马获得新记忆,以及与较老的突触竞争而表现遗忘。上述药物基于激活Notch信号通路,促进NSCs增殖和分化,改善学习和记忆能力。
4 AD相关MAPK信号转导通路
4.1 MAPK组成与调控丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)是一种丝氨酸/苏氨酸蛋白激酶,能够通过响应各种细胞外刺激来处理和调节细胞特性,其能磷酸化蛋白质中丝氨酸或苏氨酸,并发挥调节细胞存活、分化、增殖和凋亡等重要作用。如今在哺乳动物细胞中,已发现了几种不同的MAPK,包括p38 MAPK、JNK、细胞外信号调节激酶(ERK 1/2)和ERK 5/BMK-1。其中p38α(通常简称为“p38”)是第一个被鉴定的p38 MAPK亚型,并且首先被认为是应激诱导的激酶,可被脂多糖(LPS)和炎性细胞因子激活。MAPK信号通路转导由3个层次顺序的激酶依次激活,首先4种不同的MAPK途径通过小G蛋白RAS激活,活化的RAS通常会刺激RAF,进而激活MEK 1/2,最后激活ERK 1/2,但也可能使MEKK 2/3磷酸化,从而诱导另一种由MEKK 2/3到MEK 5再到ERK 5组成的MAPK级联的激活。与其他MAPK途径类似,JNK 1/2/3途径通过顺序为Rho到MEKK 1到MKK 4/7进行的蛋白激酶磷酸化激活。最后一个MAPK途径则由RAC激活TAK1再激活MKK 3/6,最终激活效应物蛋白p38 MAPK[16]。在中枢神经系统中p38 MAPK的激活与Tau蛋白过度磷酸化、ROS产生和积累、促炎因子释放、树突棘数量减少、记忆障碍、神经细胞凋亡等相关[17];JNK的激活与Aβ的生成和沉积、Tau蛋白过度磷酸化、神经炎症等相关[18];ERK的激活与神经细胞的存活有关[2]。选择性p38 MAPK抑制剂SB203580(合成化合物)和选择性JNK抑制剂SP600125(合成化合物)可以降低给予Aβ25-35PC12细胞中神经炎症标志物COX-2和iNOS的表达,改善Aβ25-35诱导的神经毒性[19]。选择性ERK抑制剂PD98059(合成化合物)能抑制清心开窍方(QKF)的抗神经元凋亡作用,提高Bax和Caspase-3的表达[20]。
4.2 信号通路与ADAlvario等[21]发现,从链霉菌中分离的两种化合物链霉素A和B能够减少脂多糖刺激的BV2小胶质细胞炎性因子的释放,诱导Nrf2易位至小胶质细胞的细胞核中,并通过抑制MAPK信号通路减少了Tau蛋白的过度磷酸化,发挥调节神经炎症和神经保护的作用。Gao等[20]发现,清心开窍方(QKF)通过增强ERK1/2磷酸化,并下调p38MAPK磷酸化,显着降低Bax和Caspase-3的水平,同时上调海马Bcl-2的水平来改善AD模型小鼠学习和记忆能力,并减少海马区细胞凋亡数量。上述药物基于调节MAPK信号通路,减轻神经炎症和减少神经细胞凋亡改善学习和记忆能力。
5 AD相关NF-κB信号转导通路
5.1 组成与调控诱导型转录因子的核因子κB(nuclear factor-κB,NF-κB)是一种普遍表达且具有翻译后调节活性的二聚体分子。NF-κB蛋白家族包括NF-κB1 p50、NF-κB2 p52、RELA(也称为p65)、RELB和c-REL。NF-κB蛋白通常的存在形式为一种非活性细胞质复合物,并与κB抑制剂(IκB)家族的成员结合,IκB家族成员除了传统的IκB蛋白还有NF-κB前体蛋白p105和p100等。NF-κB信号转导的激活有两种主要途径:经典的和非经典的NF-κB信号转导途径。其中经典途径介导了经典NF-κB家族成员NF-κB1 p50,RELA和c-REL的激活,而非经典NF-κB途径则选择性激活了非经典NF-κB家族成员NF-κB2 p52和RELB。经典NF-κB途径第一步激活TGF-β激活的激酶1(TGFβ-activated kinase,TAK1/MAP3K7),然后激活IκB激酶(IκB kinase,IKK)复合物,该复合物为一种三聚体由调节亚基IKKγ/NEMO和催化亚基IKKα及IKKβ组成,进而该复合物可磷酸化IκB家族成员如IκBα、p105等,IκB家族成员被磷酸化后会进一步发生蛋白酶体降解及泛素化,最终释放典型NF-κB家族成员NF-κB1 p50-RELA和NF-κB1 p50-c-REL二聚体同时易位至细胞核,从而使附近具有NF-κB DNA结合位点的特定基因的表达。最后,NF-κB对这些基因的激活会导致特定的生理反应,例如炎症或免疫反应,影响细胞存活和增殖。非经典NF-κB途径第一步激活NF-κB诱导激酶(NF-κB-inducing kinase,NIK),然后使IKKα磷酸化,进而IKKα磷酸化p100羧基末端的丝氨酸残基,使磷酸化的p100进行选择性降解,导致非典型NF-κB家族成员NF-κB2 p52-RELB二聚体的释放和核易位[22]。小胶质细胞是中枢神经系统中重要的免疫细胞,小胶质细胞的活化会释放炎性因子并促进神经细胞凋亡,抑制NF-κB信号通路可减少其炎性因子的释放,发挥神经元保护作用[23]。多种信号级联途径如AKT、MAPK、AGE/RAGE/GSK-3β等在激活该途径中起重要作用[24]。使用选择性NF-κB抑制剂BAY11-7082(合成化合物)证明了橙皮素基于抑制该信号通路发挥减轻神经细胞氧化应激、炎症因子释放、死亡和改善认知等神经保护作用[25]。
5.2 信号通路与ADLiu等[26]发现,番茄红素可以通过抑制NF-κB p65和TLR4的mRNA和蛋白表达,减少TNF-α,IL-1β和IL-6β的产生来减轻神经炎性损伤,改善认知功能障碍。Yang等[27]发现,针刺可以抑制NF-κB信号通路的活性,减少ROS的产生和阻止Ca2+升高,还可以调节葡萄糖代谢、增强神经传递、上调G蛋白活性、降低Aβ蛋白沉积和神经元凋亡以及降低氧化应激,其中抗氧化应激作用部分与抑制NF-κB及其下游靶基因p53有关。Qi等[28]发现,益智草五味子药对(ASHP)能够抑制NF-κB/p65蛋白的核转运和IκB-α蛋白的降解从而调节NF-κB信号通路来减少神经炎症和凋亡发挥抗AD作用。神经炎症是一种在感染和伤害的背景下生理保护性反应,但神经炎症还可以导致神经元变性。AD患者中淀粉样斑块周围存在大量激活态小胶质细胞聚集,这些细胞会产生如NO、ROS、蛋白酶、黏附分子及促炎细胞因子等神经毒性分子[23]。上述药物基于抑制NF-κB信号通路减少炎症因子释放和氧化应激来减轻神经炎症和神经细胞凋亡,改善认知功能障碍。
6 对策与展望
AD是高度复杂的神经退行性疾病,当前,全球有4 700万人患有痴呆症,估计到2050年将增加3倍多(约1.31亿),关于AD发病机制并未阐明,目前停留在假说阶段,有胆碱能神经元假说、Aβ毒性假说、Tau蛋白假说、线粒体损伤假说、神经炎症假说等,上文提到的药物及调控作用见Tab 1,截止2018年12月3日的FDA官方文章表明,FDA已批准了五种用于治疗AD的药物(多奈哌齐、加兰他敏、卡巴拉汀、他克林、美金刚胺,见Tab 2),其中四种都属于胆碱酯酶抑制剂,途径较为单一。
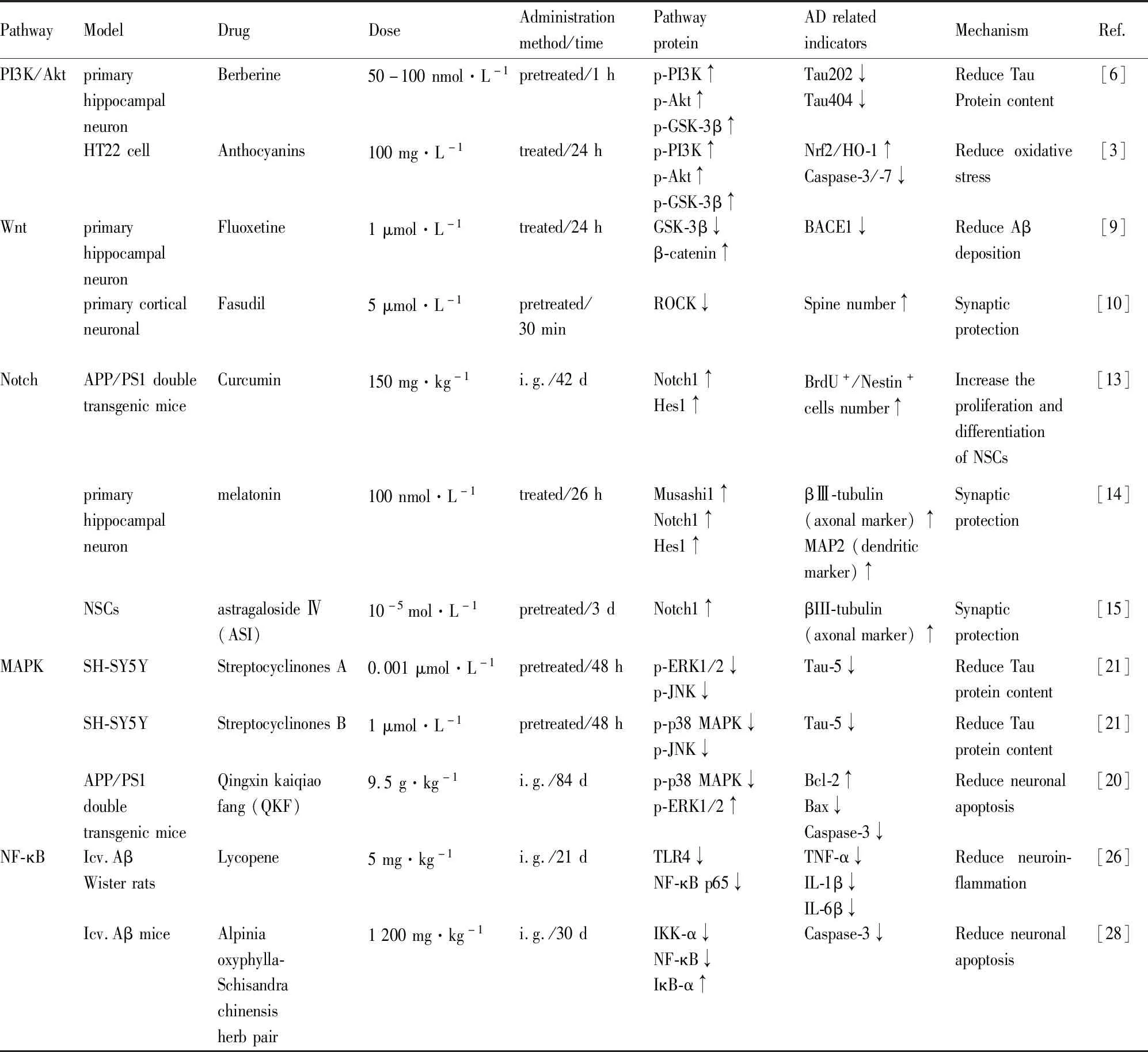
Tab 1 Pathway mechanism and indicators of drug intervention
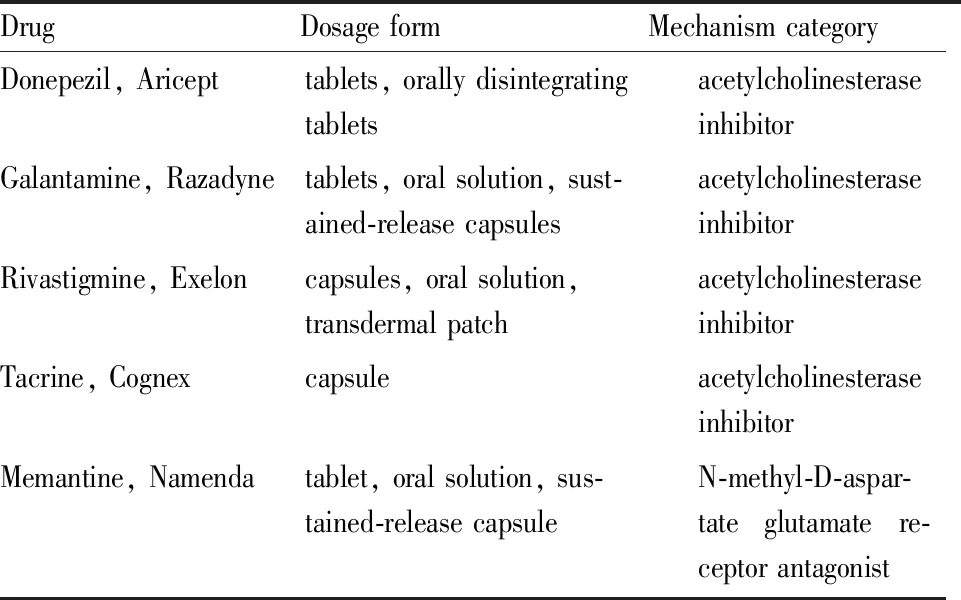
Tab 2 Five drugs approved by FDA
其中针对Aβ蛋白的药物主要作用于Aβ的生成和清除。截至2020年6月9日Cognition公司的药物CT1812进入Ⅱ期临床,其作用机制为阻止AβO与突触上的sigma-2受体结合,sigma-2受体在神经元的生长和再生中发挥重要作用,减少突触受到Aβ毒性蛋白的损害[29],提示我们在研究抗AD药物时可以关注突触保护方向,其中Wnt和Notch信号通路都与突触有关,针对Wnt通路我们可以从其关键蛋白寻找潜在新药,如Dkk1蛋白抑制剂等,Notch通路中Notch蛋白的水解切割需要多种酶的参与其中包括γ分泌酶的参与,而Aβ的生成也需要γ分泌酶的参与,使用γ分泌酶抑制剂时可以减少Aβ的生成,但同时Notch信号通路也会受到抑制导致突触损害,因此我们可以寻找基于激活Notch信号通路的突触保护药或者探寻具有减少Aβ生成而不影响Notch信号通路激活的选择性γ-分泌酶抑制剂。Tau蛋白的过度磷酸化与GSK-3β蛋白的激活有关,而GSK-3β与PI3K/Akt、Wnt、NF-κB多条信号通路有关,因此我们可以利用激活PI3K/Akt信号通路(PTEN抑制剂、Akt激活剂等)抑制GSK-3β减少Tau蛋白磷酸化,同时该通路的激活还会减少神经细胞的凋亡,或者发掘GSK-3β抑制剂,减少Tau蛋白磷酸化同时激活Wnt信号通路,抑制NF-κB信号通路,分别发挥突触保护和减少神经炎症损伤效应。随着研究的深入,现学者发现AD的病理特点并不仅见于Aβ沉积和Tau蛋白过度磷酸化,血脑屏障的改变也是神经退行性疾病的早期标志,而有研究发现AD的主要遗传因素就是载脂蛋白E4(Apolipoprotein E4,ApoE4)的高表达,且该因素与血脑屏障破坏有关,并通过MAPK信号通路增加APP的转录和Aβ蛋白的合成[30],提示我们可以开发潜在MAPK信号通路抑制剂,降低ApoE4高表达人群的患AD风险。
目前,与之相关的AD发病机制假说主要为BACE1和APP 改变,致使Aβ沉积干扰突触信号进而引起Tau蛋白过度磷酸化和小胶质细胞激活的神经炎症,其中APP形成Aβ过程中需要的γ分泌酶也同样参与Notch信号通路生理过程,该信号通路与NSCs的增殖与分化有关,上调该通路可调节神经发生、改善突触可塑性进而改善记忆和认知能力;Wnt信号通路是正常的突触功能维持所必须的通路,上调该通路可预防AD淀粉样斑块造成的突触损伤;上述两条通路的共同调节维持了突触的正常结构和生理功能。PI3K/Akt、MAPK、NF-κB信号通路主要与神经炎症和神经元死亡有关,其中PI3K/Akt、MAPK信号通路主要影响神经细胞存活,上调PI3K/Akt信号通路和抑制MAPK信号通路可减少神经细胞凋亡;NF-κB信号通路主要影响小胶质细胞促炎因子释放,抑制该通路可减轻神经炎症,但炎症关键蛋白NF-κB受到多通路如PI3K/Akt、MAPK综合调控,激活MAPK和 PI3K/Akt信号通路均能上调NF-κB蛋白的表达,与Tau蛋白过度磷酸化密切相关的GSK-3β蛋白与Wnt、PI3K/Akt、NF-κB信号通路均有关,系统关系图见Fig 1,但部分机制仍未阐明,如何通过单一或复合调控上述通路发挥潜在治疗AD的作用有待进一步研究。本文阐释了信号通路与AD的部分关系与关键蛋白网络,提示调节信号通路的关键因子或靶蛋白可能为治疗AD的潜在重要路径,可为抗AD新药的研发提供新思路。
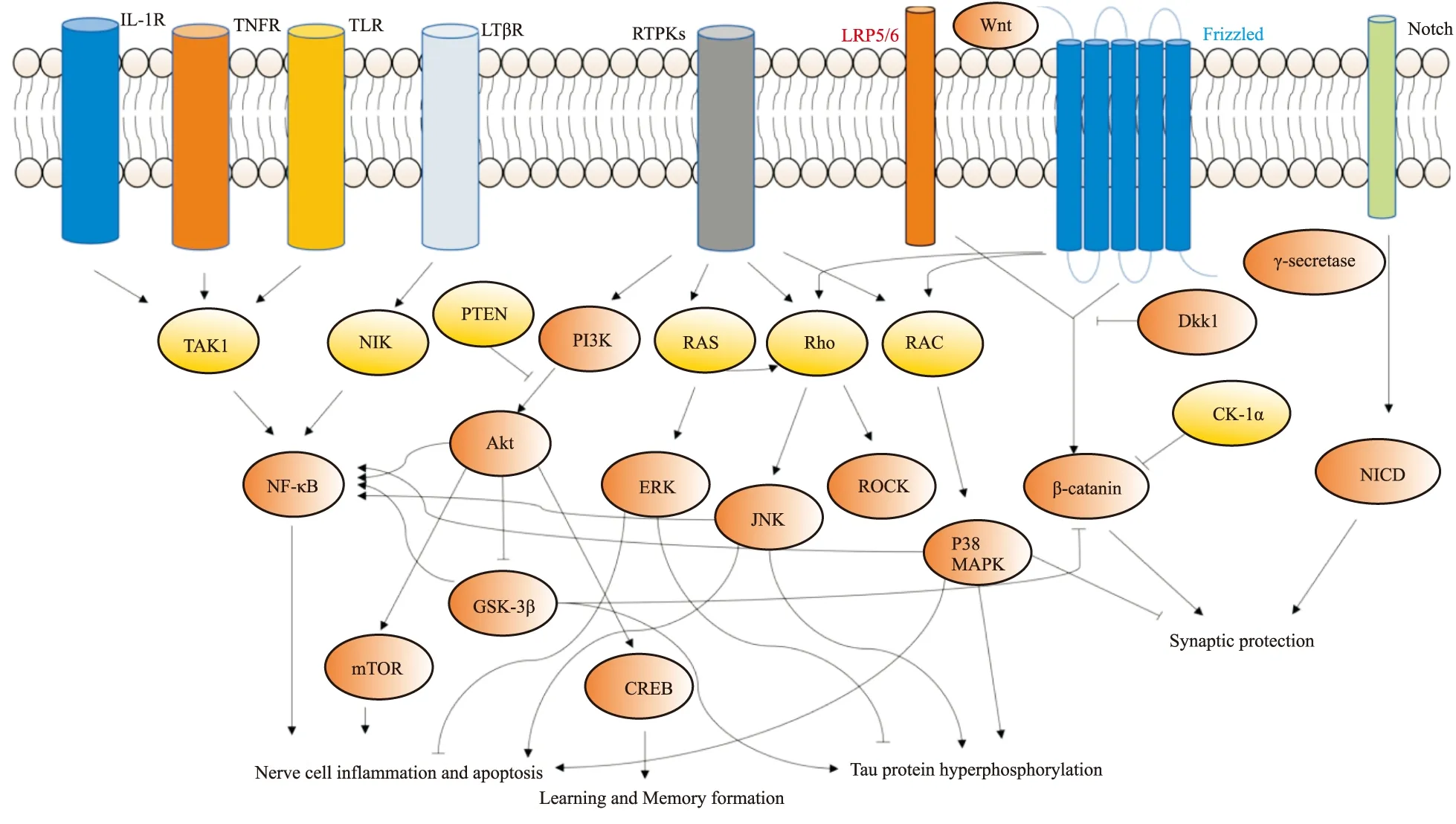
Fig 1 AD-related signaling pathways
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2019年6期)2019-08-13
天然产物研究与开发(2018年7期)2018-08-21
分析化学(2017年12期)2017-12-25
上海农业学报(2017年3期)2017-04-10
中国医药生物技术(2015年4期)2015-12-26
安徽医科大学学报(2015年9期)2015-12-16
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
