
Amyloid beta peptides,tau protein acetylation and therapeutic strategies for treating Alzheimer’s disease:a review
2021-02-07 04:56:14SanjibGuhaRajalakshmiSubramaniyam
Life Research 2021年1期
Sanjib Guha,Rajalakshmi Subramaniyam
1University of Rochester,Department of Anesthesiology & Perioperative Medicine,New York 14642,USA.2AU-KBC Research Centre,Chemical Biology and Nanobiotechnology Laboratory,Anna University,Chennai 600044,India.
Abstract Alzheimer’s disease (AD) is the most common progressive neurodegenerative disorder.It is often lethal and currently lacks a satisfactory therapy.The disease has a specific neuro-pathological profile:accumulation of proteinaceous deposits in the brain - amyloid plaques (containing β-amyloid peptides) and neurofibrillary tangles which are accumulation of a profusion of long stringy tangles of proteins called tau.Between the two highly recognized AD hypotheses,amyloid beta (Aβ) peptide aggregation and accumulation play a significant role and are considered as an important mechanism of AD pathology.Aβ is a proteolytic product of amyloid precursor protein and genetic studies supported the relevance of Aβ in AD pathogenesis.A large number of small molecules were studied for their ability to inhibit Aβ-aggregation in oligomer form or after fibrillization.However,the protein-misfolding process has certain setbacks which are inevitable due to the different morphology of protein.In recent years,it has been demonstrated that tau also plays a central role in pathogenesis of this disease.Moreover,abnormal post-translational modifications of tau,in particular,increases in acetylation at specific sites likely contribute to the toxicity of tau.Although it is evident that tau with these aberrant post-translational modifications likely facilitates neurodegeneration,the precise cellular mechanisms by which tau compromises neuronal function remain unknown.In addition,much remains to be learned about new interventions that might be developed to prevent or reduce the negative impact of tau posttranslational modifications-related damage.This review article addresses the key roles of amyloid beta and tau protein in AD as well as the possible therapeutic agents that can reduce the toxic levels of both the proteins,and thus providing beneficial effect for the AD patients.
Key words:Alzheimer’s disease,Amyloid beta peptide,Tau acetylation,Therapeutic strategies,Disease models
Background
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder persuaded by progressive decline of memory and cognitive functions,and changes in behavior and plasticity [1].Plaques and tangles are at the heart of AD pathogenesis.The extracellular aggregates otherwise known as neuritic plaques are constituted with amyloid beta (Aβ) peptides whereas the intracellular aggregates also known as neurofibrillary tangles are composed of microtubule-associated protein tau [2].Research upon protein aggregation has shown its significance for highlighting diverse physiological phenomena enabling scientists to understand the pathogenesis of many diseases [3].As discussed,two very important protein aggregation which need to be noticed in the case of AD is amyloid beta and tau protein modification [4].Common theory is that once these pathologic proteins accumulate they block synaptic connection among neurons,which leads to severe neurodegeneration and eventual death of neurons [5].Ultimately,it causes severe cortical and hippocampal shrinkage,and thus affecting nearly all the functions of the brain [6,7].Nevertheless,the precise mechanism still remains elusive.In this review,we will give more inputs on the mechanism of action of these two proteins and the potential therapeutic strategies involved in the cure of AD.
Occurrence of Aβ
Aβ can be found in lung and kidney with a relatively higher concentration in the brain.Aβ has been found both intra- and extracellularly whereas a large portion is seen to be in the brain (both intra- and extracellularly)[5].Total Aβ is distributed in an AD brain at a concentration of about 31 mg/g,fifteen times that in age-matched non-AD brains.Cerebrospinal fluid levels of Aβ (1-42),800 pg/ml are considerably higher than in plasma,about 20 pg/ml [8,9].It is necessary to understand the chemistry and the biology associated with Aβ peptides and tau protein,to gain further insight into this disease.
Human brain is the most remarkable and multifaceted organ,weighs about three pounds where nerve cells connect with one another via a tiny structure called synapse,which triggers the release of a chemical called neurotransmitter.AD completely collapses the neuronal transport function of the central nervous system.More than 26 million people are affected worldwide by AD,where this number is expected to reach over 81 million by 2040 [10].To date there is no effective treatment to combat AD and therefore prevention of neurodegeneration processes is the most urgent social and scientific imperatives.
It has been found that the culprit behind AD could be the formation of amyloid beta oligomers which appear years before the plaque development in the brain.The Aβ peptide is a product from sequential proteolysis of amyloid precursor protein (APP) by β- andγ-secretases[11].The genes which influence Aβ are - APOE,APP,PSEN1,and PSEN2 [12-14].ε4 polymorphism of APOE is responsible for a late onset of AD [15].This polymorphism causes increase in Aβ aggregation and decrease in Aβ clearance.Genetic mutation to PSEN1(on chromosome 14) and PSEN2 (on chromosome 1)gene either cause an increase in the proportion of Aβ (1-42) or enhance the aggregation into neurotoxic oligomers/protofibrils [16].APP is a glycoprotein that comprises 770 amino acids with two alternatively spliced exons,serine protease inhibitor domain of the Kunitz type (56 amino acids) [17] and 19 amino acids[18].At amino acids 700-723A a single transmembrane domain is present.The Aβ includes 28 plus the first 12-14 residues of the transmembrane domain.A second cleavage within the transmembrane region by gamma secretase produces a mixture of Aβ components,with Aβ (1-40) and Aβ (1-42) predominates.
Peptide sequence and chemistry
Aβ,a 39- to 43-residue peptide cleaved from the Cterminal region of a much larger protein is a hydrolysis product of APP catalyzed by cellular secretase.This peptide contains 39-43 amino acid residue in which Cterminal is hydrophobic and N-terminal is hydrophilic domain with high affinity metal binding sites that modulates peptide aggregation and toxicity [19,20].Monomeric Aβ forms oligomer Aβ through interaction between self-recognition site and hydrophobic Cterminal region.Aβ oligomers aggregates to form protofibrils which in turn forms fibrils (composed of β sheets).Fibrils helps in the accumulation of Aβ oligomers through fragmentation and secondary nucleation.Aβ oligomers are potential toxic species.The toxicity of Aβ oligomers arises due to their interaction with membrane receptors and disruption of intracellular processes such as mitochondrial dysfunction and signal interruption [21].
Apparently,increased levels of Aβ (1-42) increase the expression and the activity of the β-secretase.Guo ZF et al.,2013 found that the Aβ has a vastly different organization in oligomers than in amyloid plaques [22].Changes in the cleavage site catalyzed by γ-secretase elicit the deviation in length of different Aβ isoforms.The direct participation of secretases in Aβ production has led to attempts in regulating their activities to control the cerebral levels of Aβ.Such endeavors,however,have presented a lack of clinical efficacy and unforeseen consequences:for instance,γ-secretase,a complex of PS1,PS2,nicastrin and anterior pharynx defective 1 has a negative impact and affects notch signaling and invoke cognitive decline [23].
Sequencing of the peptide reveals the structural significance of specific Aβ segments:(a) Aβ1-16is involved in metal coordination; (b) Aβ17-21is the selfrecognition site responsible for driving the self-induced aggregation through hydrophobic interactions; (c) Aβ25-35is indicated as the neurotoxic fragment contributing towards aggregation.
Electrostatic interactions are known to play major roles in the self-association of peptides to form aggregates [24].In order to get self-association or aggregation,peptides need to overcome unfavorable electrostatic repulsions in the case of systems which carry net charges [25].Generally,the higher the net charge the slower the aggregation and the lower the net charge the higher the propensity to aggregate.
How polyphenols interact with Aβ peptides?
Flavonoid/polyphenols have a huge impact on neurodegenerative diseases.Because of the following reasons,(i) due to their π-π bonding between the planar faces of the polyphenol structure between the aromatic residues of proteins; (ii) sp2hybridized C,O,N (C = N,imide form); (iii) hydrogen bonding occurs between peptides and the phenolic hydroxyl groups; and (iv)facile redox chemistry between Aβ and polyphenols,polyphenols can be to a greater extent modulate Aβ fibrillization [26].Figure1 shows selective polyphenols and their impact on Aβ inhibition.
Curcumin,which plays an indispensable role in Indian food habits has shown a lot of pharmacological importance as well as a lower prevalence of AD [27].It is known that the rich antioxidant availability of polyphenols is a boon to mankind.The increased intake of flavonoids and polyphenols along with intermittent fasting is strongly said to be a preventative strategy to reduce the amyloid formation and misfolding of peptides and toxicity against the development of AD [28,29].
Tau structure and function
Research over the years has primarily focused on amyloid plaques in the context of AD,whereas less importance has been given to tau and its various posttranslational modifications (PTMs),which leads to change in the structural moiety of the protein.In this review,we primarily focus on tau acetylation at specific epitopes and report how it causes a change in a beneficial protein like tau to become neurotoxic.
Tau gene and isoforms
Human tau belongs to the class of MAPs.It is soluble,natively unfolded,50-75 kDain size,multi-functional protein and is central to the pathogenesis of AD and other neurodegenerative diseases,collectively termed as tauopathies [30].Tau is encoded by the single microtubule associated protein tau gene,MAPT,located on chromosome 17q21.31,comprising of 16 exons.It has four domains:N-terminal,proline-rich,microtubule-binding region and C-terminal.Alternate splicing of exons 2,3 and 10,generates the six primary tau protein isoforms ranging from 352 to 441 amino acids in the adult human brain [31].These six isoforms differ from each other with the presence of zero,one or two N-terminal inserts (resulting from the splicing in or out of exons 2 and 3) (0N,1N,or 2N,respectively) and either three (3R) or four (4R) microtubule-binding repeats (resulting from the splicing in or out of exon 10)in the C-terminal half of tau [32,33].These microtubule-binding repeats are essential in mediating interactions between tau and microtubules.
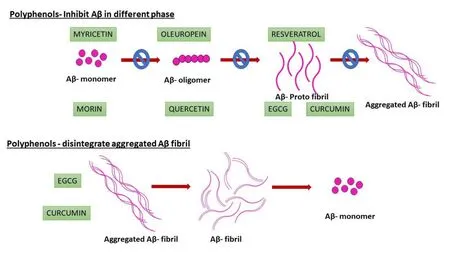
Figure1 Polyphenols that inhibit monomer,oligomer and protofibrils as well as certain polyphenols,which disintegrate the aggregated fibrils,are also shown.
Tau function
Tau is abundant in neuronal cells,especially localized in axons and at much lower levels in dendrites.Tau stabilizes microtubules,promotes microtubule assembly and most importantly regulates dynamic instability of microtubules that allows reorganization of the cytoskeleton [34,35].Other functions of tau in normal physiology include but are not limited to maintenance of healthy synapses,axonal transport,neurogenesis,neuronal polarity in development [36,37].Thus,tau protein is essential in regulating various neuronal functions in normal physiological conditions.However,the situation reverses in pathophysiology,and modified tau is one of the key causes of neuronal death in AD patients [38].Under pathological conditions,tau gets abnormally modified,gets redistributed from neuronal processes to the soma,and forms toxic oligomers or aggregated deposits,leading to the destruction of the cytoskeletal network in the dendrites[39].
Importantly,one of the main pathological hallmarks of AD is neurofibrillary tangles which are primarily composed of abnormally modified tau.Although mechanisms of toxic activity of tau are not fully recognized,it is supposed that this toxicity appears to result from soluble or oligomeric forms of tau protein,and not the insoluble neurofibrillary tangles protein [40].Tau oligomers exhibit increased,disease-associated post-translational modifications,specifically,acetylation at specific epitopes altering its turnover and function [41].
Post-translational modifications of tau
Tau acetylation
Abnormal tau acetylation plays a critical role in tauopathies and is emerging as an important physiological and pathological tau PTM [42,43].There are data indicating that acetylation inhibits the binding of tau to microtubules,enhances tau accumulation by preventing degradation and promotes the aggregation of tau in neurons [42,44].In addition,acetylation of tau contributes to the pathogenesis of AD by compromising the cytoskeletal sorting machinery in the axon initiation segment which leads to severe neurodegeneration as illustrated in Figure2.
In particular,increased expression of tau acetylated at lysine 274 (K274) and lysine 281 (K281) appears to result in mislocalization of tau to the somatodendritic compartment of the neuron from axon,destabilization of the cytoskeleton in the axon initial segment,and synaptic dysfunction [45-47].The axon initiation segment cytoskeleton is perturbed in AD and other neurodegenerative diseases.Both these epitopes are linked to cognitive decline in human AD patients [43].
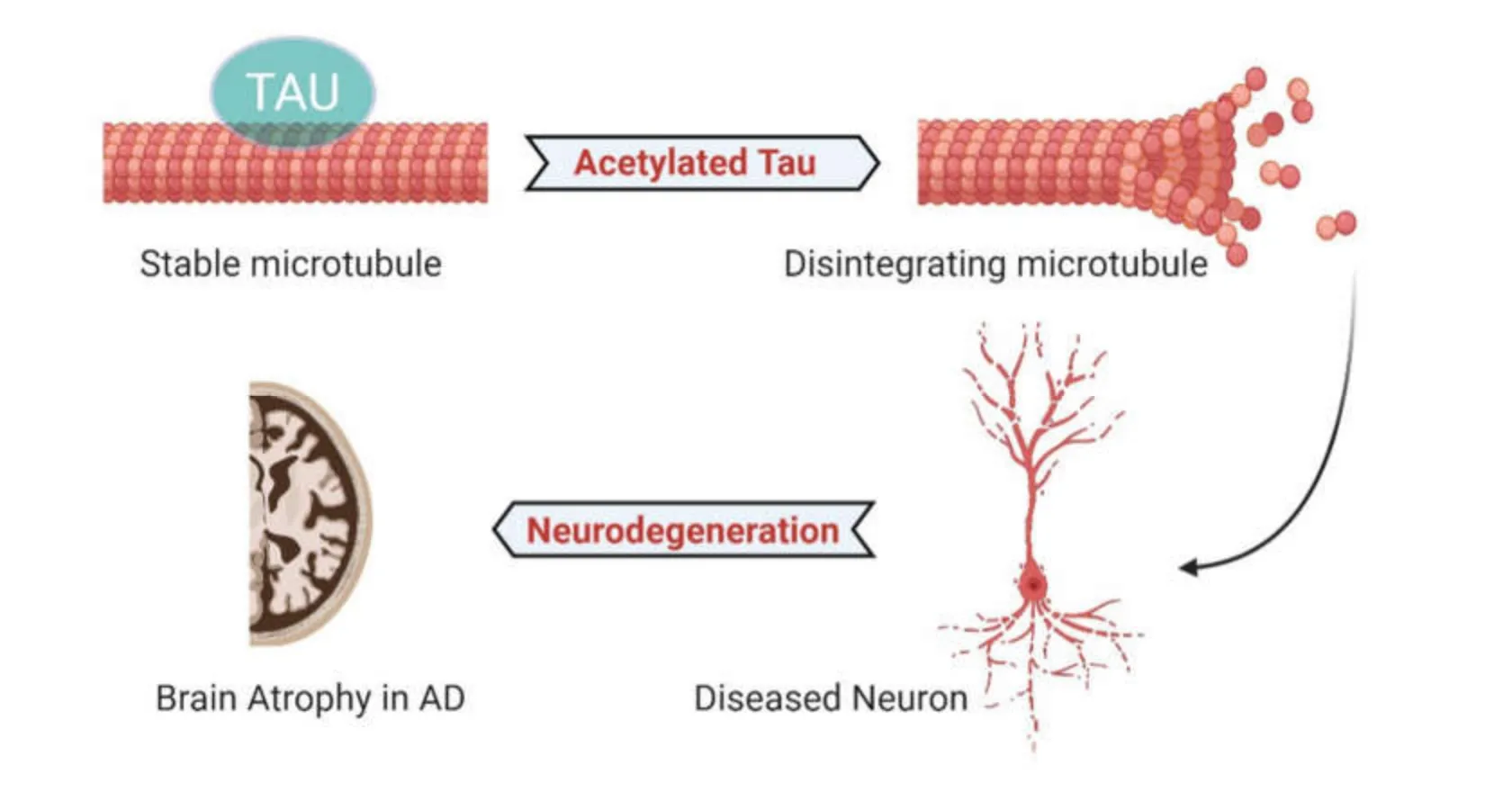
Figure2 In physiological conditions,tau normally binds to neuronal microtubule and acts as a key regulator of microtubule assembly,also helps in microtubule polymerization and neurogenesis.However,in the pathological conditions,tau is abnormally acetylated at specific epitopes,leading to disruption of microtubules,promoting the loss of dendritic spines,and initiates a key triggering event to neuronal death.
Recently,in our lab,we have usedC.elegansas a genetic model to express human tau in a defined set of sensory neurons via single-copy gene integration as a means of interrogating the role of tau PTM on mitochondria [48].Toward this end,CRISPR-Cas9 gene editing was then used to introduce AD-associated acetylation mimicking lysine → glutamine (K → Q) at the K274 and K281 position of the wild-type tau isoform [49].The pathological disease relevant tau mutants demonstrated severe neurodegeneration as well as mitochondrial morphology defects.Surprisingly,we found that wild-type tau had no effect on baseline mitophagy,and that the AD-relevant tau mutant exerted only a slight effect on its own.However,the same mutant completely suppressed mitophagy that was induced in response to low doses of a mitochondrial toxin paraquat,supporting the idea that pathological modifications of tau may sensitize neurons to stress,and that the molecular mechanism may include disrupting stress-induced mitochondrial turnover [48].
Other epitopes,such as acetylation of tau at lysines 174 and 280,have been detected in post-mortem AD,Pick’s disease,Frontotemporal Lobar Degeneration with tauopathy,and Progressive Supranuclear Palsy brain [43,50].In particular,K280 acetylated tau is elevated in patients at early and moderate AD Braak stages,and appears to hinder tau turnover,which may be critical for tau-mediated toxicity [51].Increased levels of acetylated tau block the postsynaptic signaling required for plasticity [44] as illustrated in Figure3.
However,not all the acetylation sites on tau are pathogenic and associated with tau dysfunction.For example,in vitro assays demonstrated that acetylation of the KXGS motifs in tau specifically at K259 prevents phosphorylation at the nearby site,S262 and thus maybe protective [52,53].Nonetheless,how disease relevant site-specific acetylation of tau affects its ability to regulate the neuronal cytoskeleton is not well understood,and further work is needed to delineate these mechanisms [54].Thus,in one of the following sections we discuss the different ways of how acetylation of tau can be prevented by using modulators.
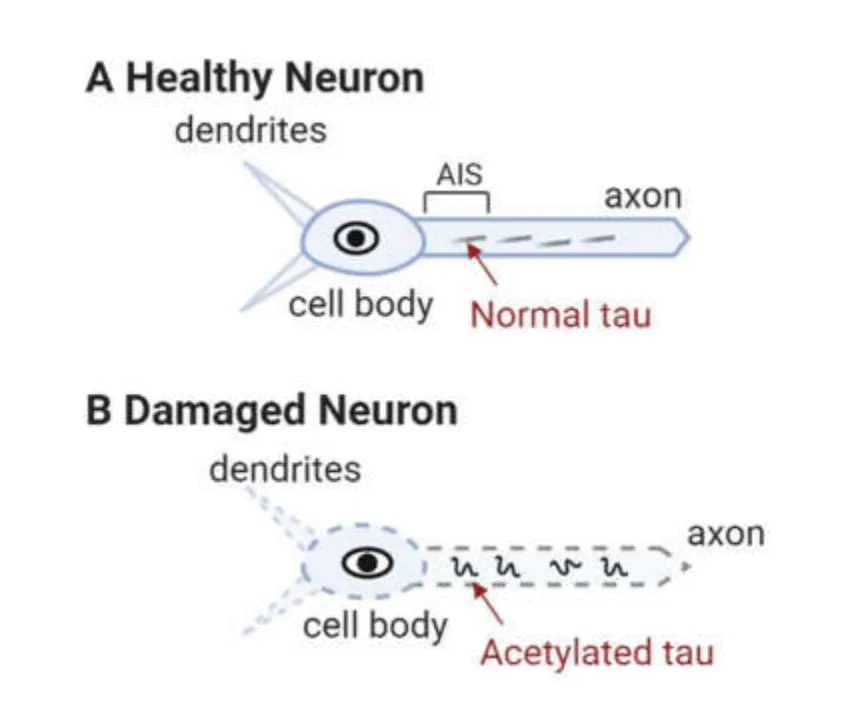
Figure3 Schematic showing how normal levels of tau helps in maintaining axonal integrity (A).In the axonal tracts,as tau gets acetylated (B),it causes severe neurodegeneration,loss of axonal transport and postsynaptic signaling is disrupted.
Other notable tau PTMs
Tau is also subject to other PTMs such as phosphorylation [55],N-glycosylation [56],glycation[57],deamination [57],nitration [58],methylation [59],ubiquitylation [60],sumoylation [61],and truncation[62].These modifications are equally important to discuss about how they potentially alters tau confirmation,ultimately resulting in altered tau function.However,they are beyond the scope of this review to be discussed and works from some of the pioneers in this field can be read here [53,63-67].
Possible therapeutic strategies to treat AD
Aβ monomers are unstable and partially folded depending on genetic mutations and external factors.Cu2+and Fe3+also induce peptide aggregation that is exaggerated at acidic pH [68].There is a large body of evidence indicating that the homeostasis of Zn,Cu and Fe,and their respective binding proteins,are significantly altered in the AD brain [69,70].Identifying small molecules/peptides which target specific site in Aβ is a valid therapeutic strategy.As we know from the literature that histidine residue presents in the Aβ metal binding site where the imidazole side chain acts as an important target to inhibit Aβ.The three significant His residues such as H6,H13 and H14 obtained out of six negatively and 3 positively charged residues have pKavalues close to physiological pH and isoelectric point is 5.5 [71].His residues are excellent ligands for copper and Aβ binds to Cu(II) with higher affinity.Cu2+binding also has the effect of lowering the pKaof a nearby amide group into the physiological range.An appreciable number of compounds have been entered into clinical trials [72].Some of them are Alzhemed (tramiprosate = 3-amino-propanesulfonic acid),Flurizan ((R)-flurbiprofen),clioquinol,Rember(methylene blue),and Caprospinol (SP-233).Alzhemed and Flurizan,both of which lowered Aβ(1-42) levels in animals,reached phase III [73].Alzhemed did not work out well whereas Flurizan shows promising results in preventing AD.Rember was targeted against tau pathology,but inhibits Aβ aggregation at the same concentration [74].Caprospinol is a compound based on the structure of cholesterol and is thought,like cholesterol,to bind to Aβ [75].
Available therapeutic options
Clioquinol,a quinoline derivative is found to be effective against Aβ peptide deposition in mice models[76].PBT2 lowers the Aβ in cerebrospinal fluid of AD patients without affecting the Cu2+and Zn2+levels.Metal chelating agents have the ability to adjust the localized metal imbalances in the brain.Recently the targeting efficacy of these metal chelators has been identified.Sarell et al.,reported that Cu2+ions show similar affinity towards amyloid β-fibrils and peptides through similar coordination mode(s) but it induces a small perturbation in the secondary structure of peptides[77].All these proved that treatment of AD with copper chelators helps to extract Aβ peptide from brain tissue by oligomer disaggregation.Figure4 explains that different coordination complexes of copper (II) helps in the inhibition of protofibrils.Compared to the total Aβ peptide content,the extractable Aβ peptide remains as a small fraction in the tissue [78].Based on the known and unknown gap in the treatment of AD using metal complexes,it is enlightened that copper complexes can be the better alternative with site specific target by tunable ligands as a drug for the treatment of AD.
Acetylation modulators
Tau can be acetylated by the histone acetyltransferases p300 or CREB-binding protein and deacetylated by histone deacetylases (HDAC),namely HDAC6 or NAD+ dependent sirtuin 1 (SIRT1) [79].HDAC6 inhibitors have shown to have neuroprotective effects as they prevent phosphorylation of tau on KXGS motifs,reducing the propensity of tau to aggregate and enhancing tau clearance [79].In AD mouse models several HDAC inhibitors reversed the learning and memory deficits,rescued tau hyperphosphorylation and decreased amyloid load,without observable adverse defects.Moreover,decreasing HDAC6 activity also promoted microtubule stability and rescued impaired mitochondrial transport through enhanced tubulin acetylation [80-82].In a recent study,it has been shown that simultaneous inhibition of HDACs and phosphodiesterase 5 has a synergistic therapeutic effect in Tg2576 AD mice model [83].Based on the findings from recent years,HDAC inhibitors seem to be neuroprotective in pre-clinical models and can be a promising strategy for AD.HDAC inhibitor,MS-275 has been in clinical trials for prostate cancer patients or patients with neuroendocrine tumor or with acute myeloid leukemia (NCT03829930 (phase I),NCT03211988 (phase II) NCT03552380 (phase II)NCT01305499 (phase II)),but not has been applied yet in regards to AD.
Resveratrol is a potent SIRT1 activator and is able to reduce hippocampal neurodegeneration,preventing learning impairment and decreasing the acetylation of SIRT1 substrates in p25 AD mouse model [84].Resveratrol stimulates key signaling pathways including the anti-oxidant defenses,reduction of inflammation via inhibiting nuclear factor-kB (NF-kB),AMPK activation leading to improved mitochondrial function and biogenesis through SIRT1 pathway [85,86].Human trial (NCT00678431,phase III) has shown that oral administration of resveratrol to mild to moderate thirty-nine AD subjects for 52 weeks improved cognitive decline and lowered Aβ levels [87,88].Although,this ensured that resveratrol parameters that were examined.Thus,interpretation ofis safe and well tolerated in patients but it failed to cause any significant changes in the different screening the resveratrol effects on clinical trajectories remain uncertain and is limited in clinical applications(NCT01504854 (phase II),NCT00743743 (phase III),NCT00678431 (phase III),NCT01716637 (phase I)).However,an interesting recent finding has claimed that due to intravenous delivery of neuronal mitochondria targeted anti-oxidant such as resveratrol,it has mitigated Aβ-related mitochondrial oxidative stress both in vivo and in vitro [89].These novel biomimetic nanosystems work by loading antioxidants into red blood cell membrane-coated nanostructural lipid carriers bearing rabies virus glycoprotein (RVG29) and triphenylphosphine cation molecules attached to the RBC membrane surface.The system can penetrate blood-brain barrier and also can target neuron cells and localize specifically in the mitochondria.Systemic administration of this nanosystem significantly improved memory impairment in APP/PS1 mice.Thinking ahead,drug candidates that can protect neuronal mitochondria from reactive oxygen species such as MitoQ,along with this Resveratrol-nanosystem in a combinatorial way would be of great value for sporadic AD treatment [86].
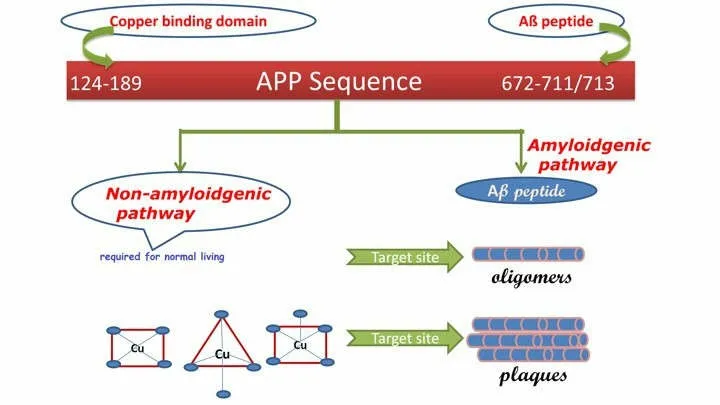
Figure4 Structural sequence of Aβ peptide from amyloid precursor protein and stepwise plaque formation from amyloidogenic pathway which different target sites are indicated.
The non-steroidal anti-inflammatory compound,salsalate,a prodrug of salicylic acid,has inhibitory activity on acetyl transferase (p300) and ameliorated tau pathology in the PS19 transgenic mouse model of frontotemporal dementia (120b).Administration of prescription drug,salsalate,after disease onset,inhibited p300 activity,lowered total tau levels as well as tau acetylated at K174,rescued acetylated-tau induced memory deficits and prevented hippocampal atrophy [47,79].These results in animal models have led to the use of salsalate in a clinical trial.This study currently at phase I (NCT03277573) is testing the effects of salsalate on cerebrospinal fluid proteins,brain magnetic resonance imaging,and cognitive (thinking and memory) tests in subjects with mild to moderate AD.Thus,acetylation modulators have recently become an interest for therapeutic development for AD patients and related tauopathies.Although,promising preclinical testing of various inhibitors of acetylation has yet to translate to cognitive benefits in human clinical trials.
Conclusion
AD is an amyloidogenic disorder characterized by the accumulation and aggregation of misfolded beta peptides.Thus understanding the structure and mechanistic pathway of monomer to fibril formation helps to rationally design and synthesize chemical compounds that could have huge impact to improve the prospects for the millions of people who suffer from this severe neuro disease.Since polyphenols have greater tendency to cross the blood brain barrier the interaction of such polyphenols with amyloid fibrils have been discussed using an analogy of curcurmin.
Interestingly,recently numerous different model systems have provided compelling data indicating that soluble tau species with disease relevant PTMs likely contribute to disease pathologies.The genetic models,especially those in which pathological tau is not overexpressed,are helping us to gain a mechanistic understanding of how soluble oligomeric tau species can cause neuronal dysfunction.In particular,the use of animal models with shorter lifespans (e.g.,worms and flies) has allowed the monitoring of the progression of pathology as function of age and its link with neuronal abnormalities.The studies presented in this review highlight how tau acetylated at various epitopes lead to disruption of microtubules,and ultimately causing neuronal death.This knowledge of cellular mechanistic insights may allow for the development of therapeutic strategies that target tau modifications to improve outcomes in AD.To conclude we have summarized the two important topics in the area of AD for the development of drugs that target Aβ as well as modified tau protein.

- Life Research的其它文章
- Science in Chinese medicine
- Artificial intelligence and the future of medicine:a multidimensional analysis
- Growth retrieval of stressed bacterial cells:logic and contradictions
- A cross-sectional study of the relationships between ethnicity and occupational classes with mental wellbeing in the UK
- Aqueous wood-ash extract of Parkia biglobosa causes reproductive toxicity in female mice
- Tear and serum electrolyte concentrations as a biomarker in primary glaucoma subjects