
数据不依赖获取的质谱数据的深度学习分析方法
2021-02-02 08:50何情祖钟传奇帅建伟韩家淮
厦门大学学报(自然科学版) 2021年1期
何情祖,钟传奇,李 翔,帅建伟,3*,韩家淮,3*
(1.厦门大学物理科学与技术学院,福建厦门361005;2.厦门大学生命科学学院,福建厦门361102;3.厦门大学健康医疗大数据国家研究院,福建厦门361102)
蛋白质谱仪的采集策略可以分为数据依赖获取(data-dependent acquisition,DDA)和数据不依赖获取(data-independent acquisition,DIA)两种.鸟枪(shotgun)法是一种典型的DDA采集模式[1].在鸟枪法实验中,质谱仪自动选择强度排名前N名的肽离子进行破碎,并记录所得的碎片离子质谱图.由于鸟枪法只选择强度排名靠前的肽段进行打碎,具有一定随机性,这使得鸟枪法检测到的肽段重复性较差.
为了解决DDA检测到的肽段重复性差的问题,2012年瑞士Gillet等[2]与AB-SCIEX公司联合开发出DIA相应的采集模式——全碎片离子顺序窗口化获取(sequential windowed acquisition of all theoretical fragment ions,SWATH).SWATH是一种典型的DIA模式,与传统的质谱数据采集方法相比,SWATH模式通过高速扫描每个荷质比窗口内所有的肽离子,并将其裂解成子离子,从而获得肽段和碎片完整的信息.相比于DDA,DIA模式具有通量高、特异性好、灵敏度高、定量结果重复性高等特点.但是DIA的质谱数据极其复杂,不同肽段的信号混合在同一张谱图内,加大了数据分析的难度.
为了分析DIA质谱数据,相关算法的开发一直是研究热点.2014年Rost等[3]提出一种文库依赖的SWATH质谱数据分析方法:OpenSWATH.该方法先使用DDA质谱数据进行数据库搜索,建立肽段的目标文库,然后根据目标文库对DIA质谱数据进行定量分析.2015年Tsou等[4]提出一款开源软件DIA-Umpire,直接分析DIA模式下的质谱数据,省去了DDA实验.在使用质谱技术研究生物学问题时,一般会制备多组样品,并且研究者主要关注不同样品间有差异的蛋白.同年,厦门大学韩家淮课题组[5]提出一种DIA的质谱数据分析方法.该方法通过计算不同组实验样品之间肽段产生的母离子和子离子信号的相关性,从而生成虚拟谱图并通过搜索引擎来鉴定肽段.同时Wang等[6]提出另一种用于DIA质谱数据的匹配分析方法MSPLIT-DIA(mixture-spectrum partitioning using libraries of identified tandem mass spectra-DIA).
此外,近两年也出现了一些基于深度学习的质谱数据分析算法.2019年Tran等[7]提出了DeepNovo-DIA算法,将深度学习和从头测序(de-novo sequencing)法结合起来,对DIA质谱数据直接进行肽段氨基酸序列的鉴定.同年,Gessulat等[8]提出了名为Prosit的基于深度学习的算法,直接从蛋白质数据库中理论预测肽段的驻留时间和子离子强度,同样摒弃了DDA实验.2020年Demichev等[9]提出DIA-NN算法,使用深度学习替代OpenSWATH工作流中打分步骤,以期发现更多的肽段和蛋白.同年,Yang等[10]提出了DeepDIA算法,同样使用深度学习从蛋白质数据库中预测肽段的驻留时间和子离子强度,其性能要强于同类的Prosit算法.
为了能够直接处理DIA质谱数据,生成虚拟谱图,不需要DDA数据进行建库,且输出文件能够与现有的肽段定性定量工作流兼容,本研究提出了基于深度学习的DIA质谱数据分析算法:Ultra-DIA.
1 Ultra-DIA算法分析流程
1.1 深度变分自动编码器

图1 3种自动编码器结构示意图Fig.1Schematic diagram of three kinds of autoencoder
自动编码器是深度学习中一种重要的无监督学习模型,主要用于数据的降维和特征抽取,它的本质是利用非线性神经网络生成的低维特征来代替高维输入.常用的自动编码器有深度变分自动编码器(deep variational autoencoder,DVAE)[11-12]、堆栈自动编码器(stacked autoencoder,SAE)[13]和降噪自动编码器(denoising autoencoder,DAE)[14]等.DVAE、SAE和DAE的结构分别如图1所示,其编码器和解码器都为多层全连接网络.其中,SAE和DAE常用的损失函数为均方差损失函数(mean square error loss, MSELoss).相较于SAE的简单结构,DAE通过随机擦除输入数据的值来学习鲁棒性更好的特征,而DVAE则进一步考虑高斯随机采样的操作,结合贝叶斯定理和KL散度(Kullback-Leibler divergence)来约束低维特征分布,从而提升了特征的表达能力和样本重构的表现.Ultra-DIA采用DVAE作为深度学习模型.
自动编码器实现的映射为:
φ:X→Z,



如图1所示,DVAE主要分成以下3大模块:编码器、高斯随机采样器和解码器.
假设存在Z的分布,但无法确定它的具体形式,只能期望在给定X的情况下推出Z分布,即p(Z|X).根据贝叶斯定理,可得:
由于p(X)表示输入数据X的未知的概率分布,所以p(Z|X)无法直接计算,那么可以使用一个可解的分布q(Z|X)去近似p(Z|X).而KL散度D可以用来描述两个分布的近似程度,其形式如下:
结合贝叶斯定理和KL散度,最终可以得到DVAE的损失L:
L=-D(q(Z|X)‖p(Z))+
EZ~q(X|Z)[logp(X|Z)],
其中E表示期望.假设q(Z|X)服从均值为μ,方差为σ2的高斯分布Ν(μ,σ2),而p(Z)服从标准正态分布Ν(0,1),那么L的第一项为:
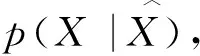
由于对Z的“采样”操作不可求导,故DVAE引入重参数化技巧(reparameterization trick),从正态分布ε~Ν(0,1)中获取随机数ε,使得采样的操作不参与梯度下降,只让采样的结果参与即可.重参数化操作为:

1.2 Ultra-DIA分析流程

图2 Ultra-DIA分析流程Fig.2Analysis workflow of Ultra-DIA
如图2所示,Ultra-DIA的分析流程分为5步.
1) 制备样品并通过AB SCIEX公司的TOF-5600质谱仪采集SWATH数据.由于原始数据为闭源的wiff格式,无法直接读取数据内容,所以需要使用质谱数据分析软件ProteoWizard包含的msconvert[15]转换工具将wiff文件转化为开源的mzXML格式文件.
2) 对DIA质谱数据进行预处理,将噪音信号通过信噪比阈值进行过滤,再对过滤后的一级质谱(MS1)和二级质谱(MS2)峰型信号进行归一化,消除数量级对后续分析的影响.
3) 使用DVAE提取母离子和子离子的特征,进而对不同子离子进行分类.经过深度学习的识别后,可以得到母离子和子离子的组合和对应的驻留时间,即可生成虚拟谱图.
4) 基于虚拟谱图,使用Comet[16-17]和X!Tandem[18]等搜索引擎对这些谱图进行数据库搜索,得到肽段的定性结果.
5) 最后使用OpenSWATH工作流对肽段的定性结果进行定量分析,最终得到肽段和蛋白的定性和定量结果.
2 结果与分析
2.1 数据情况
为了验证算法的性能,本研究制备了一个小鼠细胞的SWATH数据,梯度时间为30 min.这个数据包含100个MS1窗口,MS1质荷比范围为400~1 200,MS2的质荷比范围为100~1 800.在扫描时间上,MS1的扫描时间为250 ms,MS2的扫描时间为33 ms,一个循环的总时间约为3.6 s.
质谱数据包含3个维度的信息:质荷比、驻留时间和强度.质谱仪会采集不同时刻的谱图信号,将这些谱图按照时间排列就可以得到具有不同质荷比的离子的信号随着时间的变化曲线,称为色谱曲线.
在SWATH实验中,混合蛋白质样品被酶解后进入色谱柱进行初步分离,随后再依次进入质谱仪.因此,肽段的信号会在驻留时间方向上具有色谱峰的特征.色谱峰是判断肽段是否存在的重要依据.肽段离子的真实色谱峰呈现出高斯曲线的特征,但其一般不对称且在峰开始或者结束位置会出现干扰峰.一般而言,高信噪比的色谱峰表明此时记录到了离子信号,而低信噪比的色谱峰则是背景离子产生的干扰信号.在数据预处理阶段要针对色谱峰进行过滤,保留高信噪比的离子.
不同质荷比的母离子产生的色谱峰可能互不相同,如图3(a)所示,图中包含了5 000个母离子在1 056~2 816 s内的提取离子流(XIC)色谱曲线.不同颜色的曲线表示不同荷质比的母离子.每条曲线都可能包含多个色谱峰,每个峰都代表不同的肽段离子.在同一条色谱曲线上的不同峰表示具有相同质荷比的不同肽段.从图中可以看出,SWATH数据的MS1信号非常复杂,直接通过母离子来推断肽段序列非常困难,所以需要MS2的信息来共同鉴定肽段.而在图3(b)中,这是第50个MS1窗口内由母离子打碎产生的子离子的XIC色谱曲线.可以看出,子离子色谱曲线与母离子色谱曲线具有相似的特征,并且信噪比要高于母离子色谱曲线.
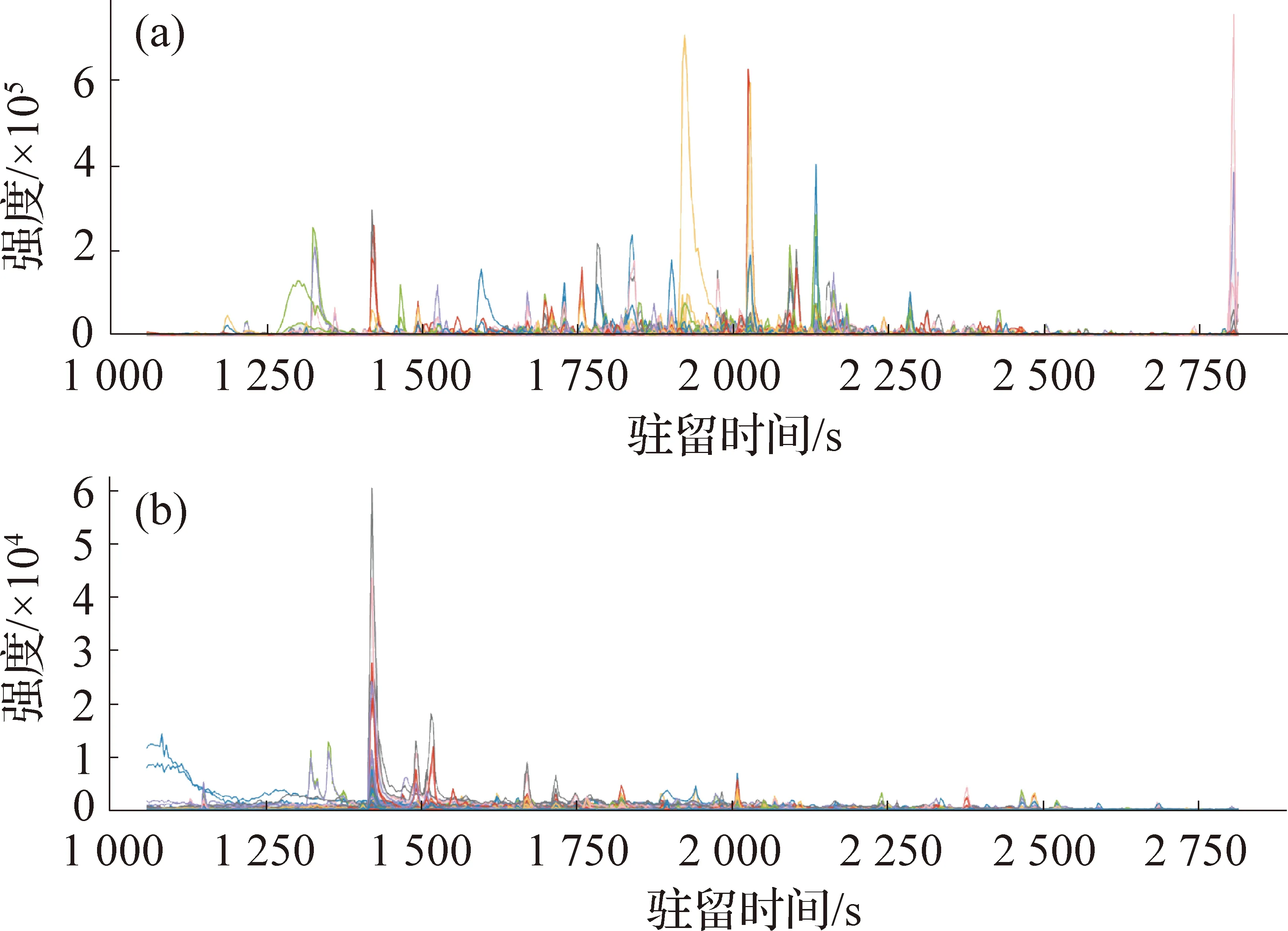
图3 典型的母离子(a)和子离子(b)的色谱图Fig.3Typical chromatograms of precursors (a) and fragments (b)
2.2 DVAE的训练
深度学习的训练是使用神经网络的重要一步,在设计好误差函数后,就可以通过优化器不断减小误差,从而达到误差最小.本研究对比了常见的6种深度学习优化算法,分别为AdaDelta、Adagrad、自适应矩估计(adaptive moment estimation,Adam)、Adamax、Nadam和随机梯度下降(stochastic gradient descent,SGD)算法[19].
如图4所示,除SGD算法外,其余优化算法的训练误差曲线在训练约25次之后开始平稳下降,并且降幅越来越小,最后趋于稳定.由于Adam算法收敛速度快,本研究最终选取它作为模型的优化器.对于Adam算法,参数设置为学习率0.001,β1=0.900,β2=0.999.
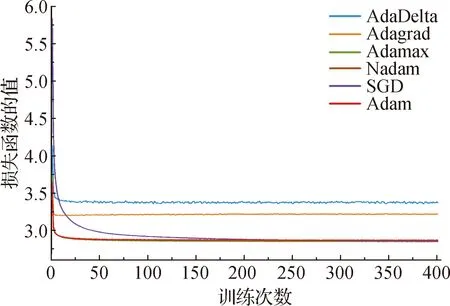
图4 训练损失曲线Fig.4Training loss curve
2.3 识别结果
2.3.1 肽段和蛋白的定性结果分析
Ultra-DIA的输出文件经过搜索引擎Comet和X!Tandem进行数据库搜索,再使用PeptideProphet、iProphet、ProteinProphet这3个软件对数据库搜索结果进行验证打分,最后使用Mayu.perl脚本计算肽段水平的1%错误发现率(FDR)的得分,用这个得分过滤肽段和蛋白即得到定性分析结果.
如图5所示,Ultra-DIA分析SWATH数据后找到的肽段和蛋白数量均大于主流软件DIA-Umpire.在表示肽段的韦恩图中,交集部分的肽段覆盖了DIA-Umpire总肽段数的78.7%;而在表示蛋白的韦恩图中,交集部分的蛋白对DIA-Umpire总蛋白数的占比达到91.7%.这说明在定性分析阶段,Ultra-DIA能够重现出主流软件的结果,其找到的肽段和蛋白可信度高.此外,在肽段水平上,Ultra-DIA单独发现的数量约是DIA-Umpire单独发现的3.6倍,在蛋白水平上达到约8倍.可见Ultra-DIA能够发现大量DIA-Umpire忽略的肽段和蛋白.

图5 肽段(a)和蛋白(b)的定性分析结果Fig.5Identification analysis results of peptides (a) and proteins (b)
此外,Ultra-DIA总共发现了4 119个肽段和1 572 个蛋白,DIA-Umpire则仅为2 662个肽段和1 016 个蛋白.在肽段和蛋白的总数上,Ultra-DIA均比DIA-Umpire增加了54.7%,可见本文算法能够找到更多的肽段和蛋白.
2.3.2 肽段和蛋白的定量结果分析
定性分析后的肽段和蛋白存在着假阳性,仍然需要进一步过滤,而过滤方式则是通过检测肽段能否定量来进行.对于SWATH数据,能够定量的肽段和蛋白才是研究人员所关注的.
本研究将定性分析后生成的谱图库和SWATH数据输入给OpenSWATH软件.OpenSWATH通过比较每个肽段对应的一组子离子色谱峰的相似度,对肽段进行评分;紧接着再通过PyProphet软件对OpenSWATH输出的评分结果进行综合,最终过滤子离子相似度低的肽段,并计算出通过筛选的蛋白数量.通过筛选得到的肽段和蛋白即是可定量的肽段和蛋白.
如图6所示,Ultra-DIA在肽段和蛋白的定量层次上,对DIA-Umpire的覆盖率分别为79.4%和94.0%,相较于定性结果都有所上升.这说明在定量层次上,Ultra-DIA能够重现出DIA-Umpire的更多结果.同时,Ultra-DIA独自发现的肽段和蛋白数量也远大于DIA-Umpire,这些被DIA-Umpire忽略的蛋白中可能隐藏着某些新蛋白.Ultra-DIA共发现3 830个肽段和1 349 个蛋白可以定量,而DIA-Umpire只发现2 373个肽段和820个蛋白可以定量.Ultra-DIA在肽段总数上比DIA-Umpire多61.4%,在蛋白总数上多64.5%.
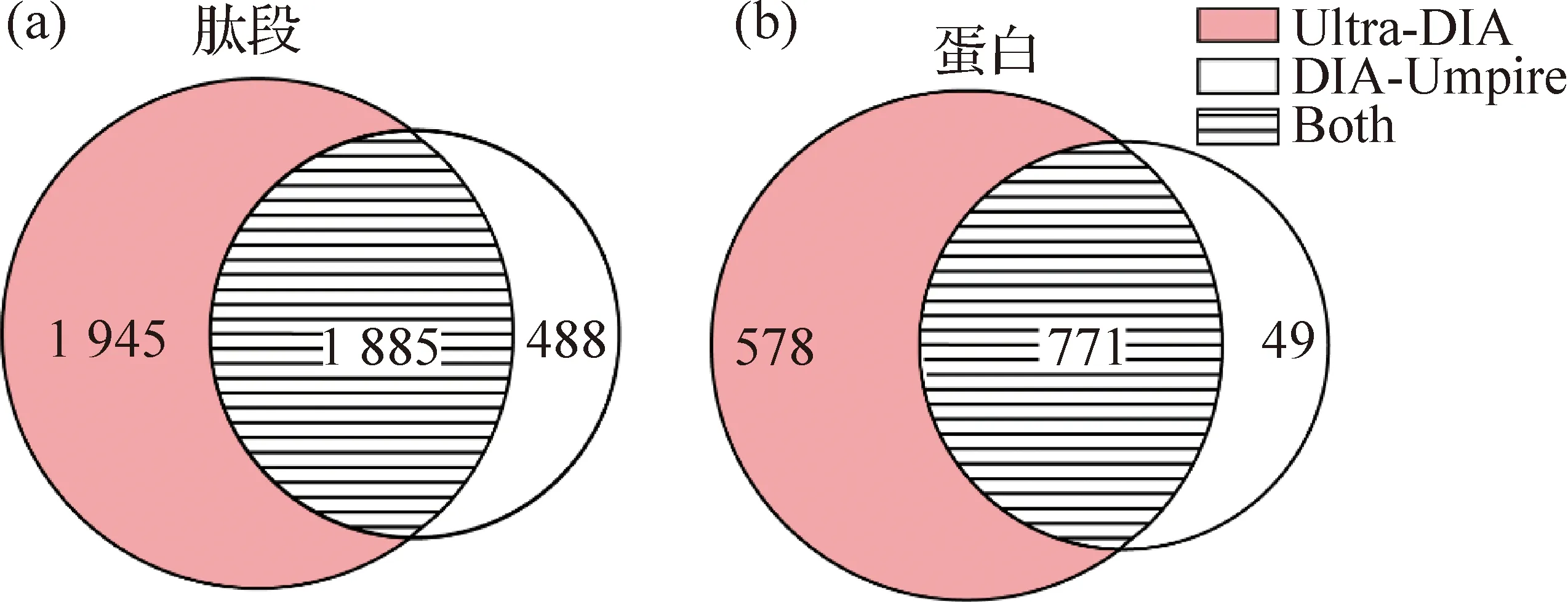
图6 肽段(a)和蛋白(b)的定量分析结果Fig.6Quantification analysis results of peptides (a) and proteins (b)
对比定性分析结果,Ultra-DIA有93.0%的肽段通过了定量筛选,而DIA-Umpire只有89.1%的肽段通过了定量筛选.同时,Ultra-DIA发现的蛋白有85.5% 可以定量,而DIA-Umpire发现的蛋白只有80.7%可以定量.这说明Ultra-DIA不仅找到了更多的肽段和蛋白,且其假阳性率更低,可信度更高.
图7展示了肽段的电荷分布,Ultra-DIA和DIA-Umpire找到的肽段的电荷具有相似的分布,都是二价最多,三价次之,四价最少.这样的分布符合仪器的设置和工作原理,说明Ultra-DIA找到的肽段可信度高.
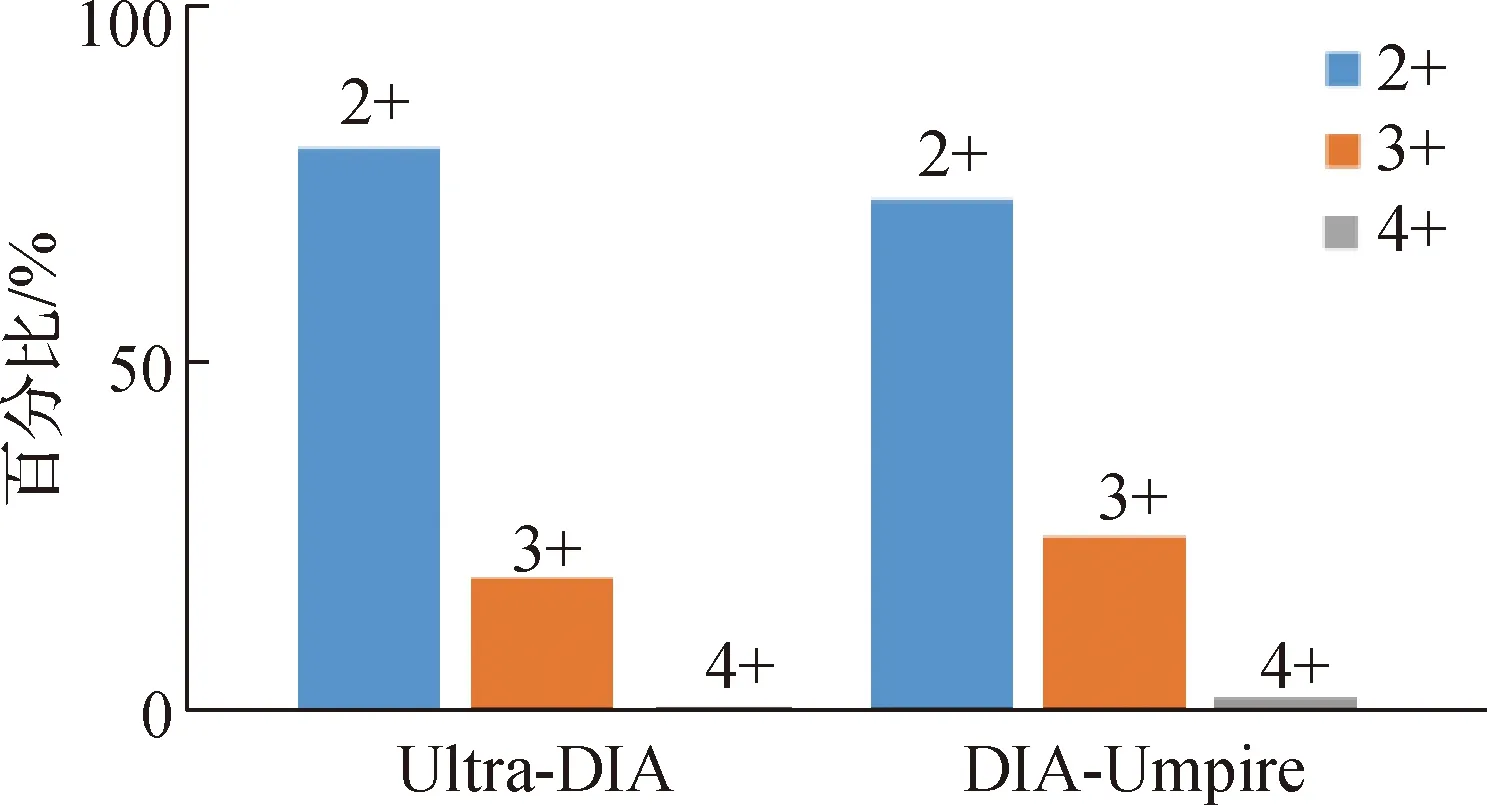
图7 肽段电荷分布Fig.7Peptide charge distribution
2.3.3 蛋白定量结果的丰度比较
在蛋白组学的研究中,样品中的低浓度蛋白一直是关注的重点.许多关键蛋白或者新蛋白的浓度都很低,其质谱信号往往被噪声淹没,强度甚至接近噪声.因此,发现的蛋白浓度越低说明算法的性能越优越.
OpenSWATH的定量结果与蛋白的真实浓度成正比.本研究将OpenSWATH对蛋白信号的定量结果取以10为底的对数,绘制蛋白信号强度的分布直方图,用来比较两种算法发现的蛋白浓度分布.
如图8所示,DIA-Umpire发现的蛋白以较高浓度居多,而Ultra-DIA则可以发现更低浓度的蛋白.这说明在分析相同数据的情况下,Ultra-DIA发现关键蛋白或者新蛋白的能力要强于DIA-Umpire.

图8 蛋白信号强度分布Fig.8Protein signal intensity distribution
2.3.4 3种自动编码器之间的比较
本研究对比了DVAE、SAE和DAE这3种自动编码器在测试数据上的肽段鉴定结果.对于SAE和DAE,还对比了MSELoss和二分类交叉熵损失函数(binary cross entropy loss, BCELoss)对结果的影响.
如图9所示,DVAE鉴定到的肽段数量最多,其次是采用BCELoss的DAE,因此在Ultra-DIA中本研究采用DVAE作为深度学习模型.此外,采用BCELoss的SAE和DAE鉴定到的肽段均多于采用MSELoss的SAE和DAE,这说明采用BCELoss提取到的特征更好.
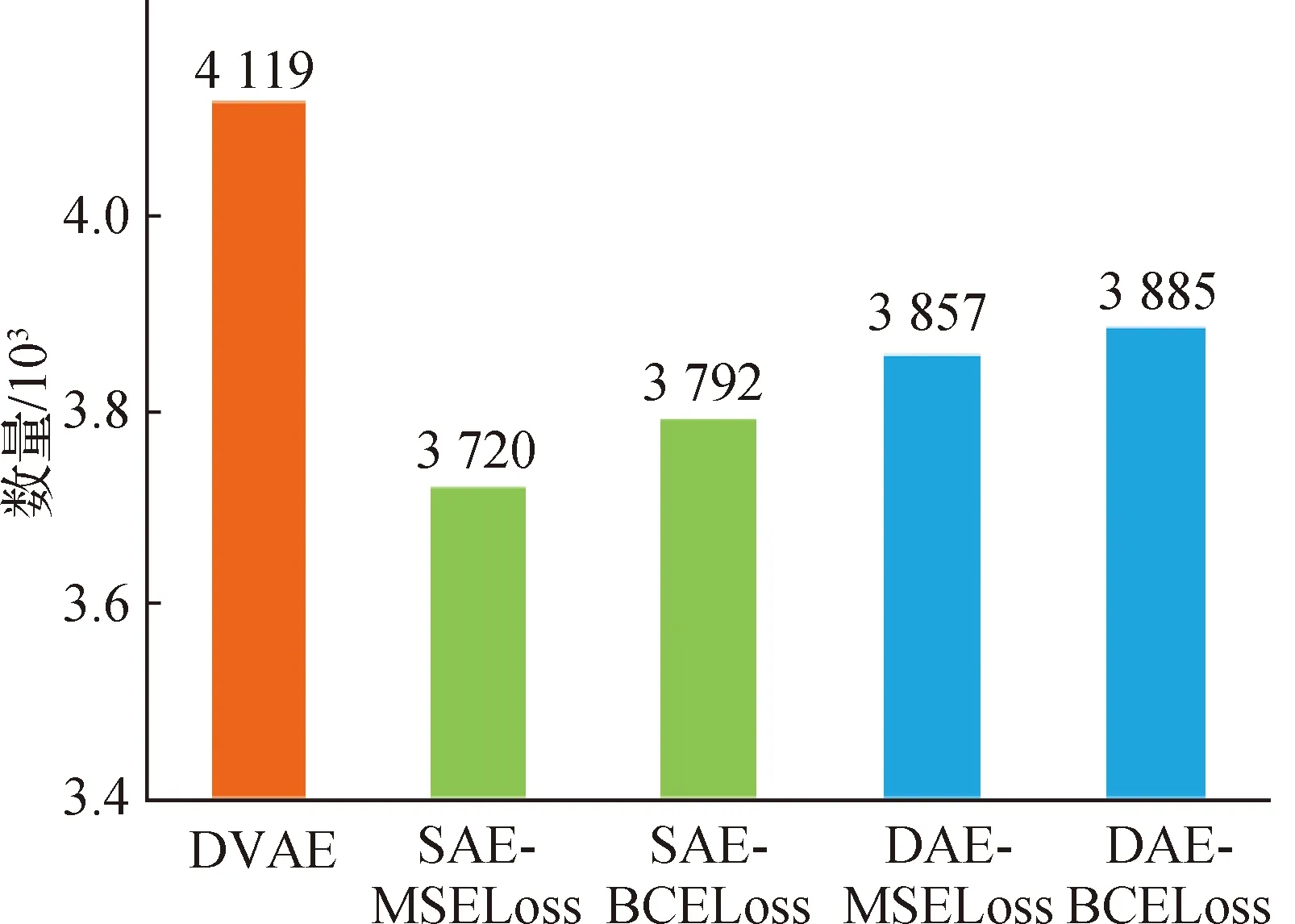
图9 不同自动编码器的肽段鉴定结果Fig.9Identification results of peptides by different autoencoders
3 结 论
本研究针对DIA数据,提出了基于深度学习的算法Ultra-DIA.该算法先对DIA数据进行预处理,然后使用深度DVAE分析DIA数据后生成虚拟谱图,再对虚拟谱图使用Comet和X!Tandem等搜索引擎进行数据库搜索,得到肽段和蛋白的定性分析结果和谱图库,最后使用OpenSWATH分析谱图库并对肽段和蛋白进行定量.
在测试数据集上,Ultra-DIA的性能要优于DIA-Umpire,但仍有许多可以改进的地方:1) 从质谱仪工作原理出发,对数据进行进一步的预处理;2) 神经网络的结构仍未达到最优,还可以设计更多不同类型的网络,通过对比最终找到的肽段和蛋白来确定哪种网络结构最合适;3) 神经网络的超参数仍未找到最优,需要不断测试;4) 测试数据的梯度时间较短,可以考虑使用不同梯度时间的数据来综合检验算法性能.
猜你喜欢
传感器世界(2022年4期)2022-08-05
中学生数理化·高一版(2022年4期)2022-05-09
现代仪器与医疗(2022年1期)2022-04-19
锻压装备与制造技术(2021年5期)2021-11-13
食品安全导刊(2021年21期)2021-08-30
现代仪器与医疗(2021年2期)2021-07-21
科学技术创新(2021年5期)2021-03-17
——编码器
演艺科技(2020年7期)2020-08-13
中国外汇(2019年22期)2019-05-21
分析化学(2019年3期)2019-03-30
