
可穿戴长时程心电记录仪临床应用的有效性和安全性研究
2021-01-31 12:36解放军总医院第七医学中心100700王晓明
首都食品与医药 2021年2期
解放军总医院第七医学中心(100700)王晓明
《中国心血管病报告2016》摘要中指出,心血管病在居民疾病构成总死亡率达40%以上,为我国居民的首位死因,目前中国心血管病患病率仍处于持续上升阶段[1]。越来越多的研究表明,心率失衡是冠状动脉疾病、心力衰竭患者死亡率相关的危险因素之一[2][3],日常生活中对心率进行有效控制和密切监测,对治疗冠状动脉疾病和心力衰竭具有非常重要的意义。心源性猝死多是由心脏原因引起的突发恶性心律失常所致,快速室性心动过速和心室颤动占75%以上[4]。许多心脏性猝死事件发生前常有晕厥或晕厥前兆症状,晕厥是指一过性脑血流低灌注而导致的突发、短暂性意识丧失,特点是发生迅速、一过性、自限性并能够自主恢复[5]。鉴于引起晕厥的原因较多,发作的突发性、发作时间不确定性等特征,临床上对不明原因的晕厥患者所做的检查,有时其病因检出率不理想。但是临床疑似心源性晕厥患者,监测有无心律失常发生、能否有效控制心率变化对确定晕厥病因及诊断,均需要长时间心电监测的结果证实。
1 资料与方法
1.1 受试者选择 入选标准:①受试者自愿参加试验并签署知情同意书;②年龄18~75周岁,性别不限;③符合动态心电图检测适应证;④能够与研究者良好交流并遵照试验要求者。排除标准:①对电极材料过敏或有严重过敏史;②无法同时符合试验和对照两套仪器;③1周之内计划进行超声、CT、MRI、发射性检查的患者;④妊娠或哺乳期妇女以及近期准备怀孕者;⑤试验前6个月有酒精或药物滥用史者;⑥有精神疾病患者;⑦1个月内参加过其他临床试验者;⑧研究者认为因其他原因不适宜参加本次临床试验者。
本试验选择了在两家医院需要进行动态心电图检查的门诊或住院受试者。共纳入了受试者60例,每位受试者均同时使用试验器械和对照器械,故试验组60例,对照组60例。本研究以符合方案数据集(PPS)和全分析数据集(FAS)作为评价的主要分析数据集。有一个受试者自行退出试验,未完成试验要求的数据采集。两组FAS和PPS均为59例。见附表1。
1.2 临床有效性评价指标
1.2.1 室性节律相关指标:①室性总数;②单个/单发室早;③成对室早数;④室速总数;⑤二联律总数;⑥三联律总数。
1.2.2 室上性节律指标:①室上性总数;②单个/单发室上早;③室上性成对数;④二联律总数;⑤三联律总数。
1.3 安全性评价指标 记录试验中发生的所有不良事件(例如,局部红肿、瘙痒、过敏等),并判断是否与试验产品相关,同时计算不良事件/反应发生率,计算公式如下:不良事件/反应发生率=(不良事件/反应发生次数)/(试验入组例数)×100%。
1.4 所采用的统计分析方法及评价方法
1.4.1 统计学设计、方法和分析规程 所有的统计检验均采用双侧检验,P≤0.05将被认为所检验的差别有统计意义。对试验组与对照组的各项数据进行统计分析,进行双侧,取a=0.05水平,若P<0.05,则两组数据的差别具有统计学意义(是有显著性差异);若P<0.01,则两组参数的差别具有统计学意义(有极显著性差异)。采用等效区间法评价相关性能指标。试验组与对照组数据均差95%,可信区下限大,采用等效区间法评价相关性能指标。试验组与对照组数据均差95%,可信区间下限大于等效区间的下限,同时试验组与对照组数据均差95%,可信区上限小于等效区的上限,则等效性成立。
安全性评价指标:试验组和对照组的不良事件发生率采用X2检验或Fisher精确概率法进行检验,详细描述各组病例出现的全部不良事件的具体表现、程度及其与器械的关系。

附表1 入组病例及安全性、有效性分析数据集

附表2 室性节律相关指标(FAS)

附表3 两组医疗器械各项室性节律相关指标结果的比较(FAS)

附表4 两组医疗器械室性节律相关指标差值的95%CI(FAS)

附表5 室上性节律相关指标(FAS)
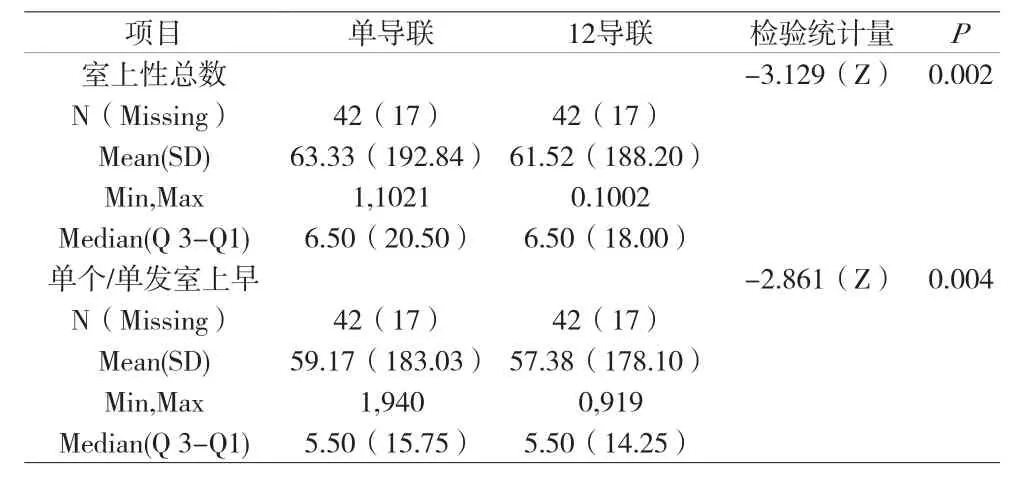
附表6 两组医疗器械各项室上性节律相关指标结果的比较(FAS)

附表7 两组医疗器械室上性节律指标差值的95%CI(FAS)
1.4.2 统计分析至少包含以下方面 ①临床试验完成情况描述:包括临床试验概括(筛选人数、入选人数、完成人数、退出/剔除人数等);②基线描述:应对所有入选受试者(FAS分析集)的基线人口统计学指标及其他相关指标等进行描述;③效果评价:应对所有入选的受试者(FAS分析集)和最终完成试验的受试者(PP分析集)分别进行统计分析;④安全性评价时,应对所有入选的受试者进行分析(SS分析集),不能遗漏所有发现的任何不良事件,对所有发生的不良事件应评价是否与所研究产品有关。
1.5 伦理情况说明 本临床试验开始前,已经获得临床研究机构伦理委员会的批准文件;研究者已准确地将知情同意书内容告知每位筛选合格的受试者并对受试者的提问进行了解答;受试者均自愿参加本项临床试验,均由受试者或其法定监护人在知情同意书上签字。在试验过程中,任何与临床试验安全性相关的问题,如临床试验方案或患者信息页的更改以及临床试验中的严重不良事件,都已及时向伦理委员会报告。
1.6 试验方案 受试仪器是海思敏自主研发的一次性长时贴片式心电记录仪(型号:HM5100T)。在两家有资质的医院伦理委员会伦理审查,并在试验前取得患者同意,签署知情同意书。采用多中心、单盲、随机和自身对照的临床试验设计方案。既符合入选标准且不符合排除标准的受试者同时佩戴24小时单通道动态心电记录仪和12导联holter,采集相应的心电图数据。隐去受试者的个人信息和使用仪器信息后,汇总数据形成临床试验报告。评价两者在测量室性节律和室上节律临床应用的安全有效性,同时评价仪器相关不良事件的发生情况。
2 结果
2.1 室性节律相关指标 指标详见附表2。
对两组室性节律相关指标(室性总数;单个/单发室早;成对室早指标;二联律总数)均进行了统计描述,均给出其各项指标的均数、标准差、最小值、最大值、中位数、四分位数间距。详见附表3。
计算两组各项室性节律相关指标(室性总数;单个/单发室早)之差(P试验组-P对照组)及其差值的95%置信区间,如果置信区间的下限均大于等效区间下限,置信区间的上限均小于等效区间上限,故可以认为试验组等效于对照组。成对室早、室速总数、二联律总数因出现情况较少,故不进行置信区间计算。详见附表4。
试验组等效于对照组。
两组各项室性节律相关指标(室性总数;单个/单个室)之差(P试验组-P对照组)的95%置信区间的下限均大于等效区间下限,置信区间的上限均小于等效区间上限,试验组等效于对照组。
2.2 室上性节律指标 室上性总数、单个/单发室上早指标试验组和对照组均同时出现的共有41例,均未出现的共有17例,试验组出现对照组未出现的1例。室上性成对指标试验组和对照组均同时出现的共有11例,均未出现的共有47例;试验组出现对照组未出现的1例。11例室上性成对数指标中,试验组数据与对照组数据完全一致,故在此不再进行分析。
二联律指标试验组和对照组均同时出现的共有1例,均未出现的共有58例。1例二联律指标中,试验组数据与对照组数据均为两次,完全一致,故在此不再进行分析。三联律指标因对照仪器不显示此指标,不在此进行分析。详见附表5。
对两组室上性节律相关指标(室上性总数;单个/单发室上早)均进行了统计描述,均给出其各项指标的均数、标准差、最小值、最大值、中位数、四分位数间距。详见附表6、详见附表7。
2.3 安全性评价 试验过程中未出现“使用过程中部件松动脱落致工作异常”、“无法启动”、“使用过程中自动关机”、“使用过程中由于机器原因出现异常中断”等情况,且试验过程中未出现过任何不良事件。
根据本试验的研究结果,单导联和12导联心电记录仪两者在室性节律和室上性节律测量等心律失常检测方面均具有较高的一致性,且使用试验产品及对照产品在整个临床试验期间未出现不良事件和其他安全性事件。
3 讨论
动态心电记录仪作为心电图学的一个重要发展和检测手段,不同于常规心电图及CCU/ICU监护心电图等心电检查方法,它是一种可以长时间连续记录并分析患者在自然生活、工作和活动状态下心电信息参数,为临床心血管疾病诊断和治疗提供重要依据。
单导联动态心电记录仪连续动态采集、记录和存储病人7天以上(含7天)的动态心电信息。动态心电记录仪通过放置在患者体表的心电电极,获取患者心脏心电活动,记录各测量点间电位差得到连续记录长时间动态心电信号。同时在室性节律和室上性节律方面具有较高的有效性和安全性,它适用于无规律、偶发性心律失常或疑似心源性晕厥患者长期自我监测,心血管疾病疑似患者及心脏病前期的筛查、诊断及长期随访,对于提高心血管疾病的检出率以及冠心病、心力衰竭等心血管疾病患者的预后具有非常重要的意义[6]。
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23
中华养生保健(2020年7期)2020-11-16
中华养生保健(2020年1期)2020-11-16
电子制作(2019年19期)2019-11-23
设计(2019年22期)2019-04-01
中西医结合心血管病电子杂志(2018年1期)2018-03-29
中西医结合心血管病电子杂志(2017年27期)2018-02-07
医学研究杂志(2015年7期)2015-06-22
中国民族民间医药·下半月(2014年2期)2014-09-26
中国社区医师(2009年18期)2009-10-26
