
极长链酰基辅酶A 脱氢酶缺乏症1 例文献复习
2021-01-20 09:18:42王璐江燕陈海燕张成祖张庆华甘肃省妇幼保健院甘肃兰州730000
医药前沿 2020年28期
王璐 江燕 陈海燕 张成祖 张庆华(甘肃省妇幼保健院 甘肃 兰州 730000)
1.临床资料
患儿,男,3 月,主因“呕吐、腹泻3 天,呻吟1 天,心跳呼吸骤停1 次”于2019 年10 月22 日收住入院。患儿于入院前3d 无明显诱因出现呕吐、腹泻,呕吐物为胃内容物,为非喷射状呕吐,大便呈黄色稀水便,量中,7 ~8 次/d,无发热、抽搐等症,家属予以“午时茶颗粒”治疗,未见明显好转。于入院前1d 出现呻吟,精神反应欠佳,就诊于当地医院,就诊过程中患儿家属发现患儿颜面部青紫,呼之不应,门诊医生立即查看患儿发现心跳呼吸骤停,立即予以心肺复苏,查血糖:0.23mmol/L,静脉推注高糖等治疗,患儿心跳呼吸恢复,为进一步诊治,急诊120 转入我院。患儿有一哥哥,出生4d 后不明原因夭折。
体格检查:体温:36.7℃,脉搏:128 次/min,呼吸19 次/min,血压87/66mmHg(1mmHg=0.133kPa),神志清,精神反应差,皮肤黏膜苍白,双侧瞳孔等大等圆,对光反射灵敏,颈软,双肺呼吸音粗,未闻及干湿性啰音,心率128 次/min,律齐,各瓣膜听诊区未闻及病理性杂音,腹软,肝脏肋下3cm,质软,脾脏肋下未触及,肠鸣音活跃,神经系统未查及异常。
入院后查血糖明显降低、中度贫血、低蛋白血症、心肌酶异常、低纤维蛋白原、电解质异常、血氨升高、肝功能异常(表1),PCT 升高,C 肽、胰岛素、甲功、肾上腺皮质功能正常。胸片提示双肺渗出性病变。心脏彩超:左室壁略增厚,心包腔积液(少量),卵圆孔未闭,房水平左向右分流,二、三尖瓣反流(少量),左右室收缩及舒张功能正常,肺动脉压未见明显异常。头颅MRI提示右室顶叶异常信号影,多考虑局灶性脑损伤。先心CTA:左位心,心室大动脉连接一致,心包积液,双肺多发节段性实变,肝脏密度弥漫性减低,呈“重度脂肪肝”表现。血串联质谱提示游离肉碱及蛋氨酸偏高,多项酰基肉碱指标异常(表2)。尿气象质谱未见明显异常。
入院后给予抗感染、纠正低血糖、保肝、输注白蛋白、纠正凝血功能异常、降血氨、营养支持等对症治疗,于入院当天凌晨再次出现呼吸心跳下降,意识模糊,立即予以气囊加压给氧、气管插管,查血气血糖测不出,给予高糖静脉推注,患儿心跳呼吸恢复。之后仍有反复类似症状发作。于11 月6 患日儿出现心跳呼吸骤停,抢救无效,宣布死亡。死亡前留取的基因检测报告提示为极长链酰基辅酶A 脱氢酶缺乏症(图1)。
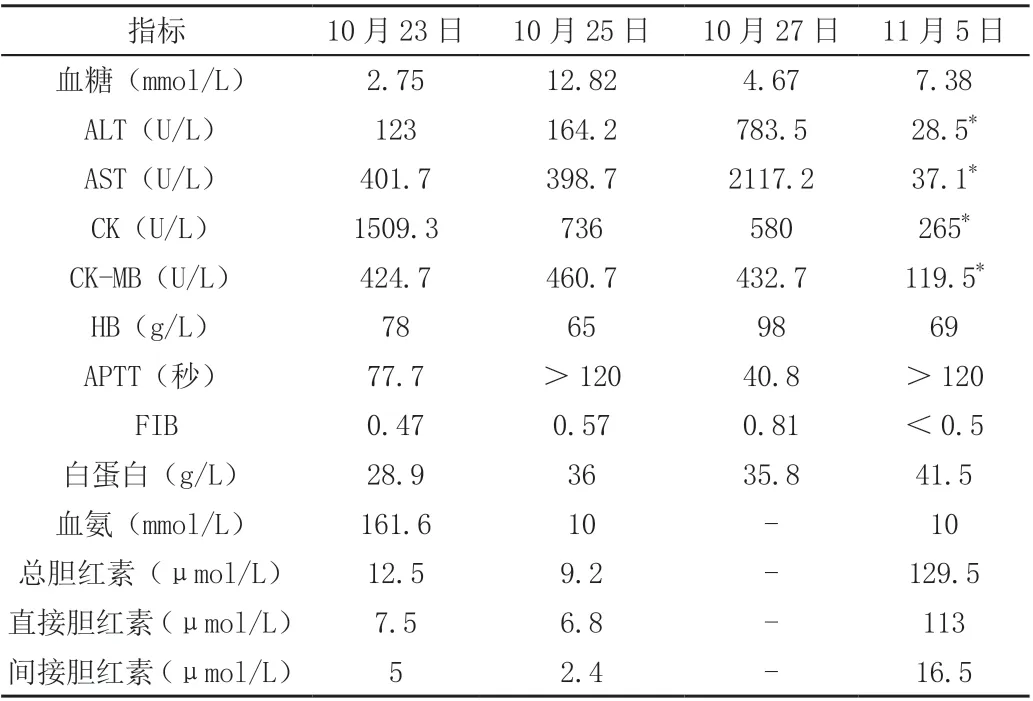
表1 主要的实验室检查
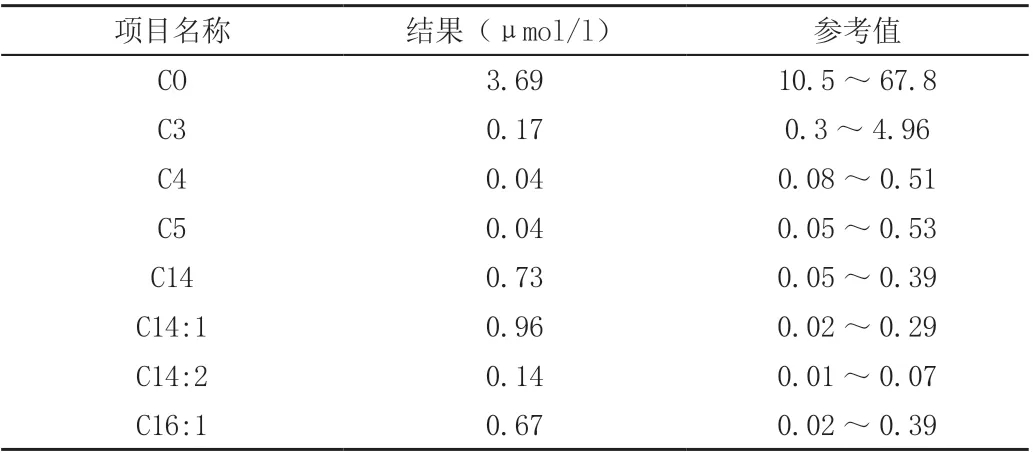
表2 血串联质谱检测结果
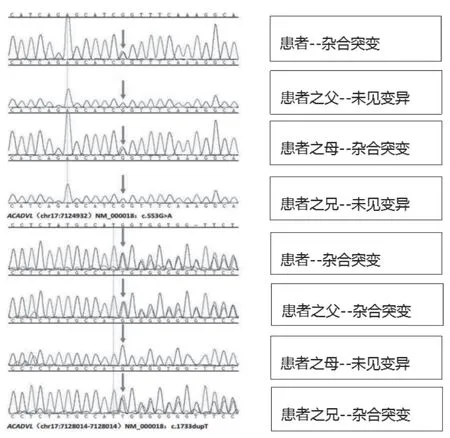
图1 基因检测结果
2.讨论
极长链酰基辅酶A 脱氢酶(very long chain acy-CoA dehydrogenase,VLCAD)可催化14 ~20 个碳原子的长链脂肪酸,是线粒体脂肪酸β 氧化第一步的关键酶,线粒体脂肪酸β 氧化是心脏、肝脏、骨骼肌等脏器组织能量供应的主要来源,当VLCAD 缺乏时可引起线粒体长链脂肪酸氧化障碍,导致机体脏器能量供应障碍而引起各种各样的临床表现[1-3]。根据临床表现的严重程度,目前将VLCADD 分为3 个亚型,即严重型(心肌病型)、中间型(肝型)、轻型(肌病型)[4]。心肌病型常为新生儿及婴儿早期发病,常有心肌受累,发病凶险,导致肥厚性或扩张型心肌病,心包积液,低血糖、呼吸困难、喂养困难、发绀、意识障碍、瑞士综合征,严重可出现新生儿猝死[5]。本例患儿为此型,表现为反复低血糖、呼吸心跳下降、心包积液、肝功能异常、颅脑损伤、心肌酶明显升高,误诊为瑞士综合征,最终因循环呼吸衰竭死亡。有研究发现低血糖及运动可诱发呼吸功能不全,病因可能为能量缺乏及呼吸肌无力共同引起[6]。该患儿反复呼吸心跳下降,可能与此有关。肝型主要为婴儿后期或儿童期发病,主要表现为反复发作性低血糖及肝功能异常,很少伴有心肌损害,经早期治疗预后较好。肌病型主要在青少年至成年发病,症状轻,常为剧烈运动,感染、饥饿、酗酒后发生横纹肌溶解,肌红蛋白尿、代谢性酸中毒、甚至出现肾衰竭,可伴有肌无力、肌痛或肌肉痛性痉挛。
新生儿筛查使用MS-MS 可确诊VLCADD[7-8],血片主要为代谢产物包括C14:1、C14:2、C14、C12:1 升高,其中以C14:1 升高最为明显[9],因此将此项指标大于1μoml/l 作为诊断VLCADD的重要代谢指标[10]。但该指标在其他脂肪酸代谢性疾病、生理性酮症也可升高,在非应激期间或已进食、静脉输注葡萄糖的患儿及临床症状轻微的患儿中也可出现假阴性。故需要行基因检测进一步明确。该患儿串联质谱提示存在多项肉碱代谢异常,但升高不明显,可能与前期患儿禁食水,静脉输注葡萄糖有关,最终行基因检测明确诊断。
VLCADD 是由ACADVL 突变所致,ACADVL 位 于染色体17P13.1,长约5.4KB,内含20 个外显子,编码655 个氨基酸多肽[11],该基因的基因型与VLCADD表现型之间存在明显相关性[12]。无义突变多见于临床症状危重的患儿,无以突变导致酶活性完全丧失,从而引发患儿的心肌病变,肝脏异常及难以纠正的代谢紊乱,错义突变多见于婴幼儿时期起病的患儿,多见于肝型及肌病型,症状轻微[13-14]。本例患儿为无义突变,临床表现危重,与之相符。ACADVL 突变谱高度异质[15],临床意义不明的新变异位点不断检出,本例患儿基因检测结果:ACADVL(NM_000018;exon7):c.553G >A;P.(G185S)(杂合)和ACADVL(NM_000018) EX:c.1733dupT;p.(M578Ifs*14);(杂合),为复合杂合突变。父亲:ACADVL(NM_000018) EX:c.1733dupT;p.(M578Ifs*14);(杂合),母亲ACADVL(NM_000018;exon7):c.553G >A;P.(G185S)(杂合)。其中C.1733dupT 为无义突变、c.553G >A(编码区第553 号核苷酸由鸟嘌呤变为腺嘌呤),导致第185 号氨基酸由甘氨酸变为丝氨酸。患儿上述变异分别遗传自父母,其父母均只有携带其中1 个杂合变异,为复合杂合突变。该患儿突变位点(C.1733dupT)为新位点突变,文献数据库未有该位点的相关报导。其父母再生育时仍有25%几率生育同样患儿,建议患儿家属再次生育时来我院遗传中心咨询,后续予以三代试管婴儿(PGT-M)助孕或者自然受孕后产前诊断,以降低出生缺陷。
VCLADD 的治疗主要包括避免进食、感染、劳累,给予高碳水化合物和低脂饮食,限制长链脂肪酸的摄入,辅以中链甘油三酯(medium-chain triglycerides,MCT),补充肉碱,短期应用可以促进酮体生成和减少空腹低血糖的发生,但过多 则会促进长链酰基肉碱的生成和蓄积,对机体产生毒性作用,其使用目前仍存在争议[16]。动态监测体内肉碱及酰基肉碱水平有助于调整治疗方案中肉碱的供给及脂肪配比,对于监测治疗效果,改善预后有极其重要的作用。动物实验表明在运动前补充酮酯对VCLADD 患者有益处,但仍需进一步临床证实[17]。
总之VCLADD 为常染色体隐性遗传性疾病,若临床出现高度可疑患儿时,如新生儿筛查试验异常提示VLCAD 缺乏;严重的早发性肥厚或扩张型心肌病,心包积液和心律不齐伴有肌张力低下,肝肿大和间歇性低血糖;严重的早发多器官衰竭;肝肿大、肝功能不全及反复的低血糖,但无心肌病,实验室检查可能有转氨酶升高或肝合成功能障碍;运动不耐症,肌肉痉挛和/或疼痛以及因剧烈运动,禁食,受冷或发烧引起横纹肌溶解导致的间歇性肌病,间歇性升高的肌酸磷酸激酶(CK)等,应及早行串联质谱检测及基因检测,及早行相关治疗,对基因诊断患者可为 VLCADD 患者家庭提供产前诊断和遗传咨询。。
猜你喜欢
种子(2021年3期)2021-04-12 01:42:22
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
家庭百事通·健康一点通(2017年3期)2017-03-22 20:13:17
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
当代化工研究(2016年2期)2016-03-20 16:21:23
中国洗涤用品工业(2016年2期)2016-02-28 19:03:17
动物营养学报(2015年9期)2016-01-07 11:29:44
应用化工(2014年5期)2014-08-08 13:10:58
高中生学习·高一版(2014年6期)2014-07-05 10:20:16
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
