
Compromised therapeutic value of pediatric liver transplantation in ethylmalonic encephalopathy: A case report
2021-01-13 09:34GuangPengZhouWeiQuZhiJunZhuLiYingSunLinWeiZhiGuiZengYingLiu
World Journal of Gastroenterology 2020年40期
Guang-Peng Zhou, Wei Qu, Zhi-Jun Zhu, Li-Ying Sun, Lin Wei, Zhi-Gui Zeng, Ying Liu
Abstract
Key Words: Ethylmalonic encephalopathy; Liver transplantation; Mitochondrial disorder; ETHE1; Pediatric; Case report
INTRODUCTION
Ethylmalonic encephalopathy [EE, Online Mendelian Inheritance in Man number #602473][1]is a rare autosomal recessively inherited disorder of mitochondrial metabolism caused by homozygous or compound heterozygous mutations in theETHE1gene, which is located on chromosome 19q13[2]. To date, more than 100 patients with EE have been reported worldwide[3]but rarely reported in China. However, based on the results of newborn screening from Jining city in 2014-2015, live-birth incidence of EE is approximately 1:25000[4]. ETHE1 deficiency results in chronic accumulation of hydrogen sulfide (H2S) and its derivative thiosulfate in multisystemic tissues such as colonic mucosa, liver, muscle, and brain[5,6]. Characteristic patients with ETHE1 disease present with (1) progressive neurological degeneration, including global neurodevelopmental delay, early-onset psychomotor regression, progressive pyramidal and extrapyramidal signs, and seizures and (2) diffuse microvasculature injury, including recurrent petechial purpura, orthostatic acrocyanosis, and chronic hemorrhagic diarrhea[6,7]. Currently, the mainstay of conservative treatment options aimed at lowering production and promoting detoxification of H2S is a methionine and isoleucine restricted diet and combined use of metronidazole (an antibiotic against enterobacterial anaerobic flora) and N-acetylcysteine (a cell-permeable precursor of the H2S scavenger glutathione)[8]. For EE patients presenting with severe encephalopathy, intravenous N-acetylcysteine infusion therapy may be more effective[9]. Since most affected individuals are compound heterozygotes resulting in unclear genotypephenotype correlation, some patients with milder courses of EE who receive combined treatments with metronidazole and N-acetylcysteine may survive into childhood and adolescence, even though they are likely to suffer acute episodes of metabolic decompensation. Kitzleret al[10]found that during an acute episode of metabolic decompensation, continuous renal replacement therapy may help to regain metabolic control in patients with EE. Despite this, the overall outcome of this devastating disorder remains poor. Progressive clinical deterioration along with psychomotor regression usually result in death within the first few years of life.
Since 2016, Dionisi-Viciet al[3]and Tamet al[11]successively performed liver transplantation (LT) in three children with EE. Although psychomotor delay remained after liver transplant, progressive improvements of neurological function and psychomotor development and dramatic amelioration of biochemical abnormalities were observed. These results suggest that LT may be a promising therapeutic approach that can improve the prognosis of EE. However, worldwide experience is still limited, and potential benefits of LT must be weighed against the potentially high risk of this procedure. Herein we describe a case of living donor liver transplantation (LDLT) for an 18-mo-old patient with EE. To our knowledge, this is the first case in Asian-Pacific countries.
CASE PRESENTATION
Chief complaints
A 15-mo-old boy with the diagnosis of EE was admitted for consideration of LT.
History of present illness
The patient was the first child of non-consanguineous parents after an uncomplicated pregnancy. He had persistent mucoid diarrhea since birth, and scattered petechiae and ecchymosis occurred in the periocular and extremities (Figure 1A). By 9 mo of age, the boy still had poor head control and no ability to sit or roll over unaided. Then, he was referred to a local children hospital. A lactose-free diet and administration of levocarnitine, benzhexol hydrochloride, coenzyme Q10, and multivitamins were initiated. However, during 3-mo follow-up, no obvious improvements in diarrhea, acrocyanosis, and developmental retardation occurred to him.
History of past illness
The patient had no other significant medical history.
Personal and family history
There was no significant personal and family medical history.
Physical examination
The neurological examination revealed significant axial hypotonia, muscle weakness, increased deep tendon reflexes, clonus, and sustained bilateral Babinski sign.
Laboratory examinations
Urine organic acid profile revealed elevated EMA at 142.53 (reference range: 0.00-6.20) and a slight increased isobutyrylglycine and 2-methylsuccinic acid at 3.61 (reference range: 0.00-0.40) and 7.99 (reference range: 0.00-6.40), respectively. Plasma acylcarnitine profile revealed elevated C4 at 2.61 μmol/L (reference range: < 1.00 μmol/L) and C5 at 1.11 μmol/L (reference range: < 0.65 μmol/L) (Figure 2). Gene sequencing analysis showed compound heterozygous mutations in theETHE1gene, a previously reported pathogenic mutation (c.375+5G>A chr19:44030348 splicing)[2]and a novel mutation (c.462T>A chr19:44015632 p.D154E) (Figure 1B).
Imaging examinations
Brain magnetic resonance imaging (MRI) by our institution revealed fronto-temporal atrophy with multiple bilateral symmetrical abnormal low and high signal intensity of basal ganglia in T1- and T2-weighted images, respectively (Figure 3).
FINAL DIAGNOSIS
The patient was diagnosed with EE.
TREATMENT
A sulfur-containing amino acids restricted diet and the combined use of metronidazole (30 mg/kg/d) and N-acetylcysteine (80 mg/kg/d) were started for the boy until we found that he was allergic to N-acetylcysteine (manifested by fever and rash), for which N-acetylcysteine was stopped immediately. Following 3 mo of combined medical and dietary treatments, urinary EMA levels decreased slightly, but remained elevated at 74.93, and values of isobutyrylglycine and 2-methylsuccinic acid rose to 5.84 and 11.31, respectively. Plasma acylcarnitine profile showed a slight decrease in C4 (2.40 μmol/L) and increase in C5 (1.42 μmol/L) (Figure 2). No obvious improvements were observed in his clinical conditions.
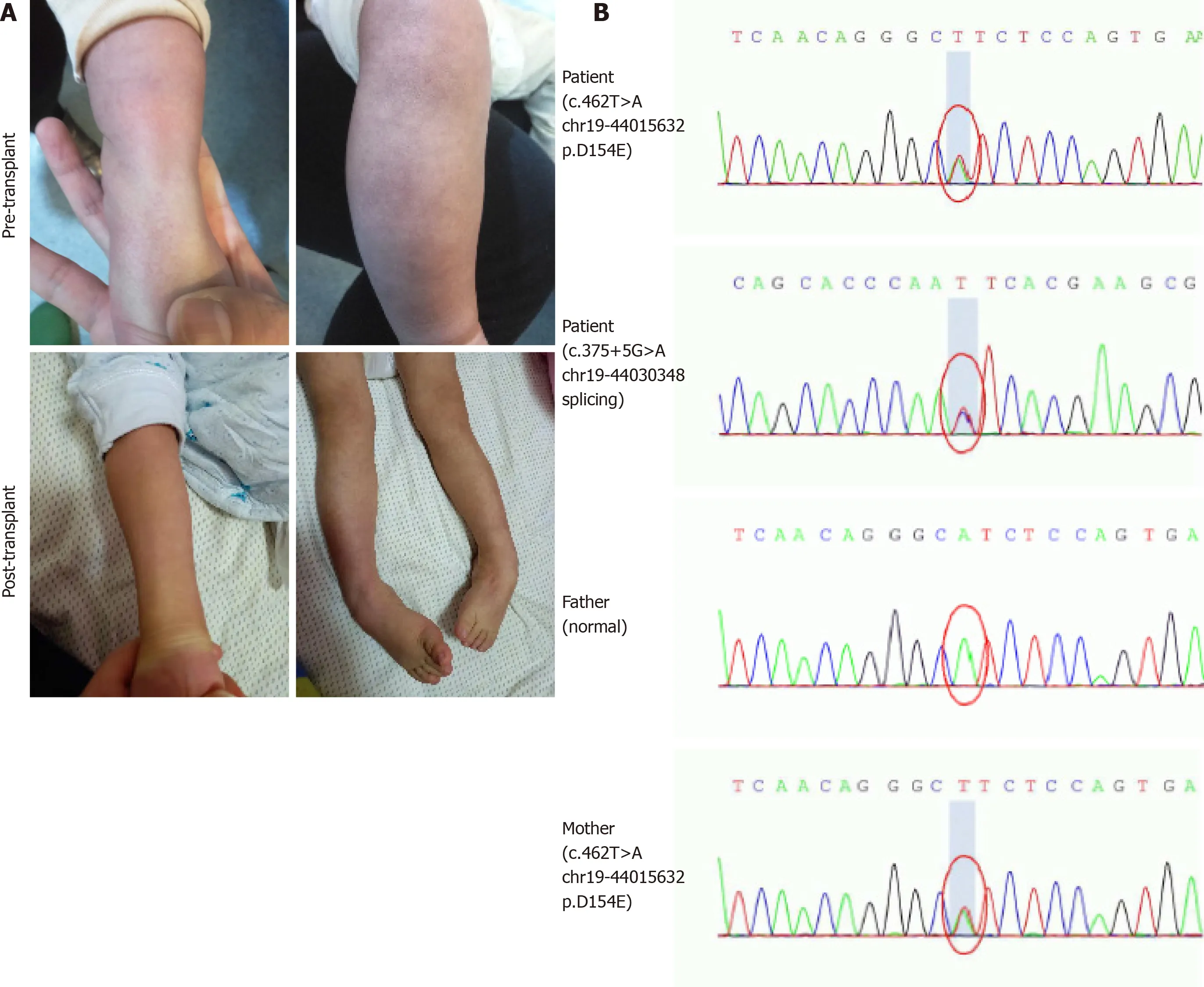
Figure 1 Clinical symptoms and gene sequencing analysis. A: The pre- and post-transplant scattered petechiae in the extremities and orthostatic acrocyanosis; B: Sequence electropherogram of the compound heterozygous mutation (c.375+5G>A and c.462T>A) in patient, heterozygous mutation in mother, and no mutation in father.
At the age of 18 mo, the boy received a LDLT with reduced-size left lateral lobe donated by his heterozygous mother under the approval of the Ethical Committee of Beijing Friendship Hospital, Capital Medical University.
OUTCOME AND FOLLOW-UP
The patient underwent a smooth operation, and postoperative course was uneventful. Immunosuppressive treatments consisted of tacrolimus and low-dose corticosteroids. Administration of metronidazole and levocarnitine was continued during the perioperative period, and metabolite levels decreased evidently. Urinary EMA, isobutyrylglycine, and 2-methylsuccinic acid levels decreased to 9.90, 0.30, and 2.41, respectively, on post-transplantation day 9. Plasma C4 increased and C5 decreased, and they did not return to normal levels (Figure 2). However, due to the recipient’s intolerance of metronidazole, the administration was privately discontinued by his mother 2 mo after transplantation.


Figure 2 Changes of pre- and post-transplant urine organic acid profile and plasma acylcarnitine profile in the patient with ethylmalonic encephalopathy.
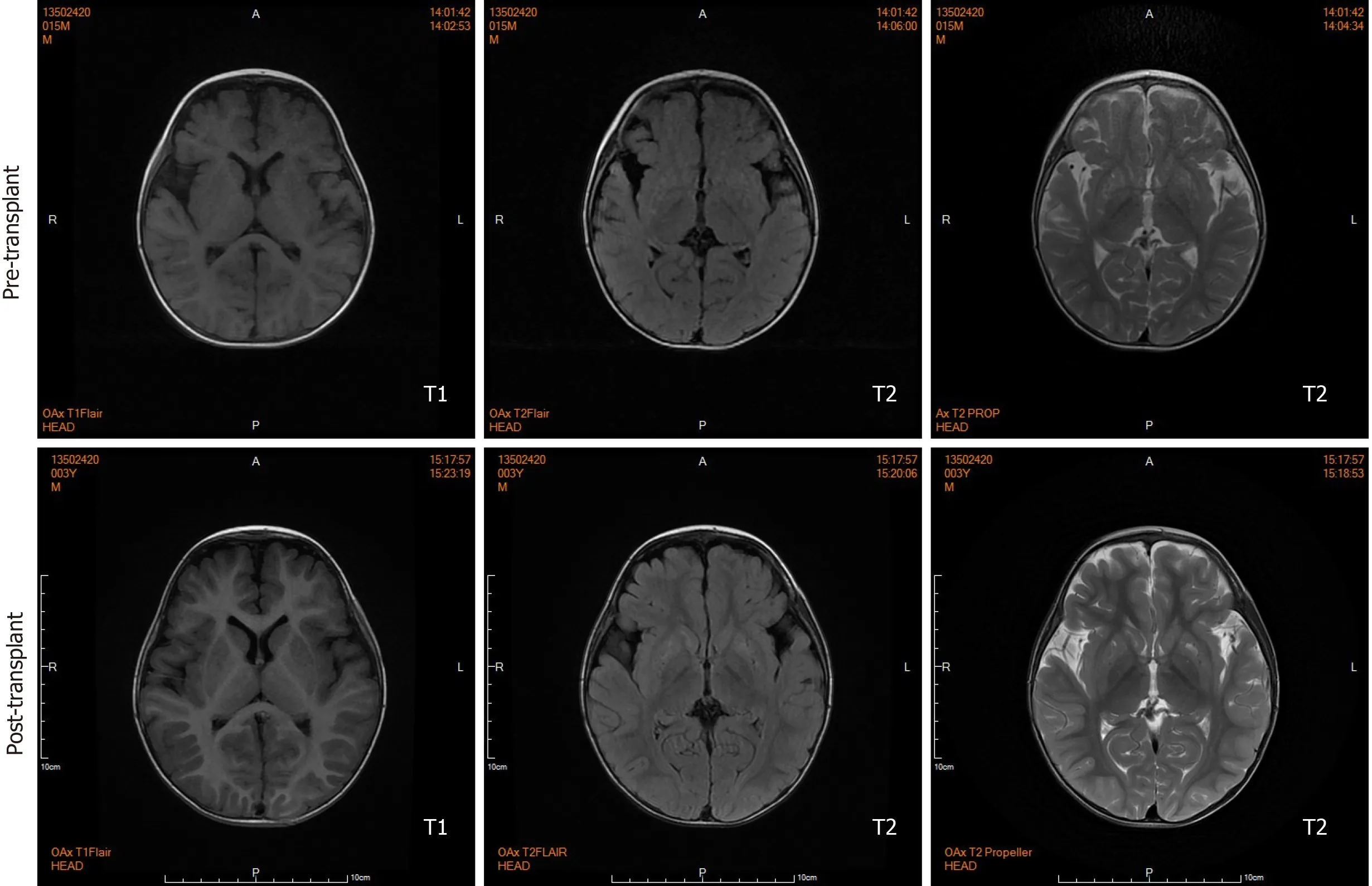
Figure 3 Pre- and post-transplant axial sections of brain magnetic resonance imaging scan. Before liver transplantation, brain magnetic resonance imaging shows fronto-temporal atrophy with multiple bilateral symmetrical abnormal low and high signal intensity involving caudate nucleus and lentiform nucleus in T1- and T2-weighted sections, respectively. Twenty months after transplantation, brain magnetic resonance imaging shows lesions remained but ameliorated.
During the 20-mo follow-up period, the patient was administered levocarnitine supplementation without a strict protein-restriction diet. Although his liver graft function remained constantly in the normal range, no remarkable amelioration of clinical conditions and neurological development was observed. The longitudinal evaluations disclosed a wild fluctuation in urinary EMA levels, far beyond the normal range. Urinary 2-methylsuccinic acid levels gradually restored to normal, whereas the concentrations of urinary isobutyrylglycine and plasma C4- and C5-acylcarnitines fluctuated markedly and still remained above the reference limits (Figure 2). Only mild amelioration of petechiae and ecchymosis was observed, and no dramatic reversion of chronic mucoid diarrhea and orthostatic acrocyanosis occurred (Figure 1). Although brain MRI suggested a certain improvement in basal ganglia lesions (Figure 3), developmental delay and neurologic disability continued. Now, at the age of 38 mo, the boy has a certain extent of head control, but is still unable to roll over, sit, stand, or walk by himself. He can just say several simple words, such as mama and papa. Meanwhile, the living donor recovered well with no postoperative complications during the follow-up.
DISCUSSION
EE is an inborn error of metabolism caused by a defect in the mitochondrial sulfur dioxygenase. The widely distributed metabolic deficit leads to significant accumulation of toxic metabolites in multiple organs. Patients with ETHE1 deficiency are characterized by a relentless progressive clinical deterioration, usually leading to death within the first decade of life. Current therapeutic approaches are most often supportive, having no remarkable impact on the natural history of EE[8,12].
Di Meoet al[13]found that intravenous injection of adeno-associated virus 2/8 carrying theETHE1gene into the livers of constitutiveEthe1−/−mice resulted in obvious amelioration of clinical phenotypes, spectacular prolongation of survival, and improvements in biochemical parameters. Based on the above findings, replacing the metabolically defective liver may help regain enzyme activity, and thus LT has been viewed as one of the most potential treatment modalities for EE. Since the first attempt in 2016, LT is emerging as a promising therapeutic option for this devastating disease[3,11]. However, the clinical outcomes of LT in our patient was not favorable. Though 20 mo have elapsed since LT, the recipient merely achieved minimal improvements in biochemical features as well as the lesions in the brain MRI without significant clinical and neurological amelioration, which compromised the promising therapeutic value of LT in EE.
Since EE is a multisystemic disease, the underlying enzymatic defect also exits in other tissues besides liver. Although the implanted liver graft containing normalETHE1gene expression could help regain missing enzymatic activity, the overall metabolic defect is only partially corrected, as extrahepatic synthesis and accumulation of toxic metabolites remain[6]. Illustrating this fact, after successful LT, postoperative levels of blood and urine metabolites among three previous recipients[3,11]and our patient did not decrease to the normal ranges. Nevertheless, cases including ours suggest that LT does have a definite role in defending against the devastating natural progression of EE. Moreover, satisfactory neurodevelopmental improvements and partial reversal of biochemical abnormalities were observed in those three liver transplant recipients. Our patient, by contrast, has not yet fully benefited from the promising therapeutic effects of LT. The detailed characteristics of these four patients are summarized in Table 1. It can be inferred that our patient’s unfavorable prognosis may be attributed to the late diagnosis of EE, late initiation of conventional treatment, pre-transplant administration of metronidazole alone, and post-transplant discontinuation of metronidazole. Furthermore, undefined genotype-phenotype correlation and the use of a heterozygous carrier donor in LDLT may also lead to the post-transplant suboptimal clinical outcomes of our patient.
The long-term clinical prognosis, especially neurodevelopmental outcomes, of liver transplant recipients depends largely on the pre- and post-transplant metabolic control. Early establishment of diagnosis and early initiation of conventional supportive management before the onset of clinical symptoms are of paramount importance as they can control the H2S overload, thereby improving clinical outcomes and biochemical markers[14]. The early clinical manifestations and neurologic deterioration arising from EE may present soon after birth[12]. It is necessary for physicians to strengthen their understanding of EE in order to make an early and correct diagnosis for these patients. After the diagnosis and before LT, combined medical and dietary treatments should be initiated without delay. It should be emphasized that only correct medical and dietary treatments, namely the combined use of metronidazole and N-acetylcysteine and a diet restricted in sulfur-containing amino acids, can help minimize H2S toxicity. Given LT cannot be performed until optimal age and weight criteria for liver transplant are achieved, conventional treatments in newborns until transplant could therefore contribute to opportune LT. The continuous administration of metronidazole and N-acetylcysteine is also necessary after liver transplant to minimize accumulation of toxic metabolites and maximize therapeutic effect of LT. Nevertheless, heterogenous genotype-phenotype correlations may influence the residual ETHE1 enzymatic activity, severity of phenotypes, and response to conventional interventions in patients with EE, which may partially explain the difference in outcomes with LT among the present several patients. In addition, although previous study suggested that the use of asymptomatic heterozygous carrier donor in LDLT for this autosomal-recessive condition was viable with satisfactory clinical outcomes[3], ETHE1 enzyme activity within the different heterozygous carrier donors’ livers may vary. However, due to the unavailability of enzyme activity measurement technology, we did not perform donor enzyme activity assay before LT. Thus, whether the compromised therapeutic effect in our patient is associated with the use of a heterozygous carrier donor remains unclear. Notably,DDLT: Decreased donor liver transplantation; N/A: Not available; LT: Liver transplantation; LDLT: Living donor liver transplantation; OLT: Orthotopic liver transplantation.there was no description of dietary restriction for the three previous liver transplant recipients. Whether a diet restricted in sulfur-containing amino acids could further improve liver transplant recipients’ clinical prognosis deserves further research.
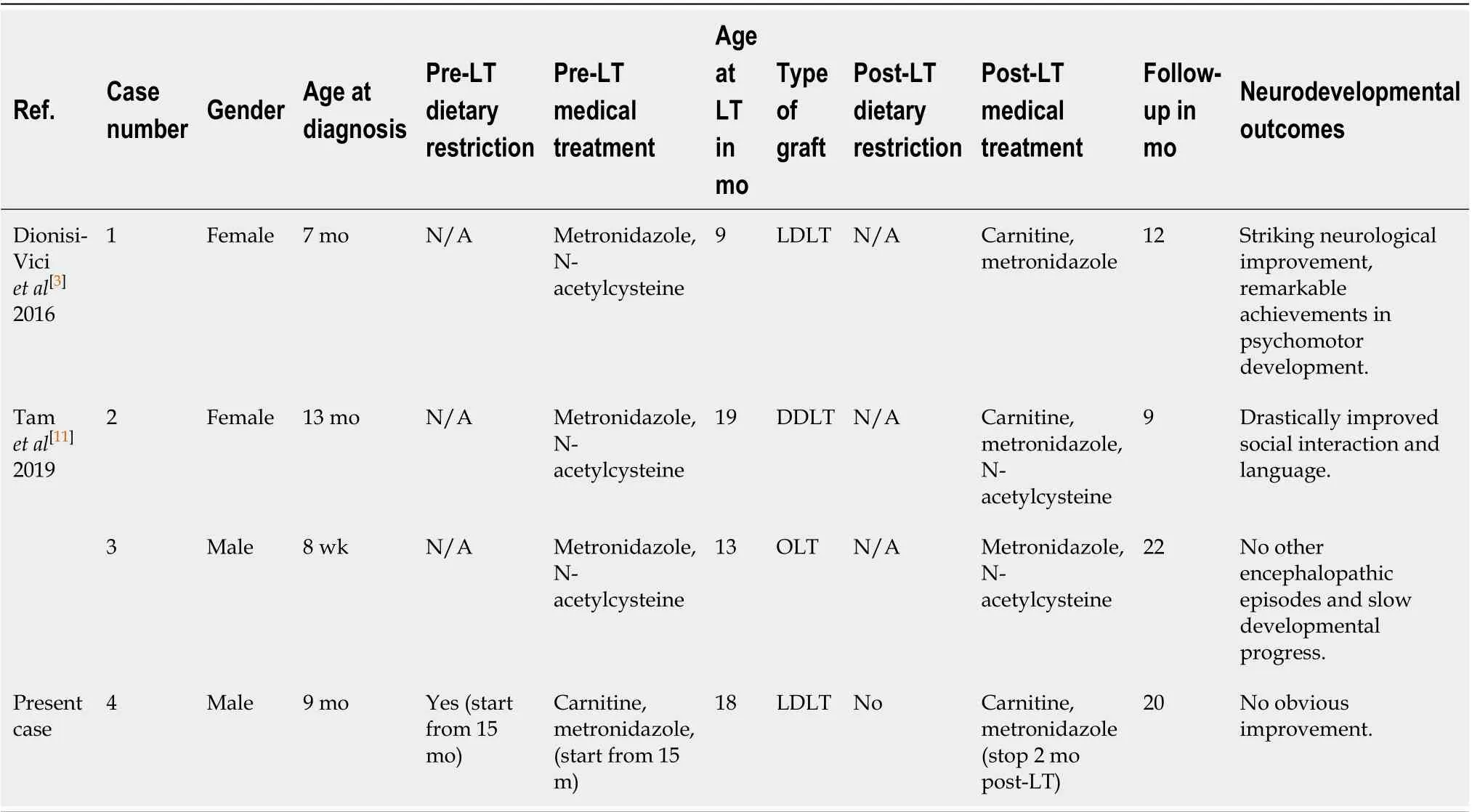
Table 1 The characteristics of liver transplant recipients with ethylmalonic encephalopathy before and after liver transplantation
Although the reversal of neurological damage still needs to be further observed in the long run, the possibility of irreversibility of neurological disability posttransplant should always be kept in the mind of doctors and patients’ guardians. Taking into account the perioperative morbidity, transplant-related complications, and long-term immunosuppression, scrupulous preoperative clinical assessment of patient’s overall clinical conditions, especially severity of neurological damage, is necessary for the evaluation of the benefit–risk ratio. More importantly, the joint participation of a multidisciplinary team including metabolism physicians, pediatric hepatologists, pediatric neurologists, pediatric transplantation team, metabolic nutritionists, and neurorehabilitation physicians, is indispensable for the long-term management of patients with EE.
CONCLUSION
LT does have a definite role in defending against the devastating natural progression of EE. Despite only a partial cure, LT should still be considered for patients with EE in the absence of other superior therapeutic options. Early establishment of diagnosis and initiation of conventional treatment pre-transplant, timely LT, and continuous administration of metabolism-correcting medications post-transplant may be helpful in minimizing the neurologic impairment and maximizing the therapeutic value of LT in EE.
ACKNOWLEDGEMENTS
We would like to thank He EH, Xu RF and Yi ZX who contributed to ultrasound monitoring in this patient.
 World Journal of Gastroenterology2020年40期
World Journal of Gastroenterology2020年40期
- World Journal of Gastroenterology的其它文章
- Cirrhotic portal hypertension: From pathophysiology to novel therapeutics
- New strain of Pediococcus pentosaceus alleviates ethanol-induced liver injury by modulating the gut microbiota and short-chain fatty acid metabolism
- Prediction of clinically actionable genetic alterations from colorectal cancer histopathology images using deep learning
- Predicting cholecystocholedochal fistulas in patients with Mirizzi syndrome undergoing endoscopic retrograde cholangiopancreatography
- Novel endoscopic papillectomy for reducing postoperative adverse events (with videos)
- Pediatric bowel preparation: Sodium picosulfate, magnesium oxide, citric acid vs polyethylene glycol, a randomized trial