
Cirrhotic portal hypertension: From pathophysiology to novel therapeutics
2021-01-13 09:34LakmieGunarathneHarindaRajapakshaNicholasShackelPeterAngusChandanaHerath
World Journal of Gastroenterology 2020年40期
Lakmie S Gunarathne, Harinda Rajapaksha, Nicholas Shackel, Peter W Angus, Chandana B Herath
Abstract Portal hypertension and bleeding from gastroesophageal varices is the major cause of morbidity and mortality in patients with cirrhosis. Portal hypertension is initiated by increased intrahepatic vascular resistance and a hyperdynamic circulatory state. The latter is characterized by a high cardiac output, increased total blood volume and splanchnic vasodilatation, resulting in increased mesenteric blood flow. Pharmacological manipulation of cirrhotic portal hypertension targets both the splanchnic and hepatic vascular beds. Drugs such as angiotensin converting enzyme inhibitors and angiotensin II type receptor 1 blockers, which target the components of the classical renin angiotensin system (RAS), are expected to reduce intrahepatic vascular tone by reducing extracellular matrix deposition and vasoactivity of contractile cells and thereby improve portal hypertension. However, these drugs have been shown to produce significant offtarget effects such as systemic hypotension and renal failure. Therefore, the current pharmacological mainstay in clinical practice to prevent variceal bleeding and improving patient survival by reducing portal pressure is non-selective -blockers (NSBBs). These NSBBs work by reducing cardiac output and splanchnic vasodilatation but most patients do not achieve an optimal therapeutic response and a significant proportion of patients are unable to tolerate these drugs. original work is properly cited and the use is non-commercial. See: htt p://creativecommons.org/License s/by-nc/4.0/
Key Words: Portal hypertension; Cirrhosis; Intrahepatic vascular resistance; Hyperdynamic circulatory state; Splanchnic vasodilatation; Portal blood flow; Non-selective betablockers; Alternate renin angiotensin system
INTRODUCTION
Cirrhosis and its complications are responsible for a large number of deaths worldwide annually. The Global Burden of Disease study in 2017 reported over 1.32 million cirrhosis-related deaths globally, which was approximately 2.4% of all deaths worldwide[1]. Almost 90% of the patients with cirrhosis eventually develop portal hypertension and this condition is a prequel to the majority of deaths in cirrhotic patients[2]. Portal hypertension can also develop in the absence of liver cirrhosis and this is termed non-cirrhotic portal hypertension[3].
The majority of complications that occur in patients with decompensated cirrhosis including the development of gastro-esophageal varices, ascites, spontaneous bacterial peritonitis (SBP), hepato-renal syndrome (HRS), hepato-pulmonary syndrome (HPS), hyper-splenism, and even hepato-cellular carcinoma, are related to the evolution of portal hypertension[4-7]. Discussion on these complications other than portal hypertension and variceal bleeding is beyond the scope of this review and the reader is referred to excellent reviews published elsewhere[8-12]. Gastroesophageal varices are the most clinically significant outcome, contributing to 25%-50% of mortality in patients because of their tendency for rupture and haemorrhage[2,13,14].
During the past few decades, many advances have been made in the understanding of the pathophysiology of portal hypertension in cirrhosis. Initially it was suggested that portal hypertension is simply a consequence of the distortion of normal tissue architecture and function in the cirrhotic liver[15,16]. However, it was subsequently shown that increased portal inflow caused by splanchnic vasodilatation also plays an important role[15,17,18].
These advances in understanding have led to the development of new therapeutic strategies. However, these new treatment options, including the use of non-selective -blockers (NSBBs), are not effective in all patients highlighting the importance of developing novel therapies that can precisely target portal hypertension in cirrhosis. Recent research suggests that the renin angiotensin system (RAS) is an important physiological system that contributes to portal hypertension and it is therefore a potential target for future pharmacotherapies[19-21]. Supporting this, many studies have shown that drugs which modulate the RAS reduce portal pressure in cirrhotic animal models and human patients with portal hypertension[22-26].
This review outlines the pathophysiological mechanisms related to the development of portal hypertension in cirrhosis and current therapeutic approaches in the management of this condition. Based on the evidence from recent advances in RAS research, in particular work on the role of the “alternate axis” of the RAS, in this review we specifically emphasize the contribution of the RAS to the pathogenesis of cirrhotic portal hypertension and how this system may be manipulated to treat this condition.
PATHOPHYSIOLOGY OF PORTAL HYPERTENSION
Changes in the pressure along a blood vessel (ΔP) result from the interaction of blood flow (Q) and the resistance (R) opposed to that flow. Accordingly, changes in the vascular resistance and/or blood flow will translate into the changes in the pressure. The same physical properties can be used to determine the factors contributing to portal hypertension. In the portal circulation, Q represents the portal venous inflow and R represents the portal vascular resistance opposed to that flow. The development of cirrhotic portal hypertension is a cumulative effect of increased portal venous resistance to the incoming venous flow due to anatomical and functional changes in the intrahepatic circulation and increased portal venous flow as a result of splanchnic vasodilatation and increased cardiac output[27,28].
INCREASED INTRAHEPATIC VASCULAR RESISTANCE
Increased hepatic vascular resistance in the cirrhotic liver is a result of both the fixed obstruction created by hepatic structural changes, and alteration of hepatic vascular tone. Vascular obliteration by the formation of scar tissue and regenerative nodules in the cirrhotic liver during tissue remodelling and scarring contributes to approximately 70% of the increase in hepatic resistance[29,30]. Activated hepatic stellate cells (HSCs) responsible for excessive production of extracellular matrix (ECM) promote scar tissue formation, thereby replacing the functional liver tissue with fibrous matrix[19,31,32]. The remaining 30% increase in hepatic resistance is governed by the contraction of activated HSCs and vascular smooth muscle cells (VSMCs) which is modulated by a range of vasoconstrictors[24,25]. The level of angiotensin II (Ang II), a potent vasoconstrictor peptide of the RAS, is elevated in the cirrhotic liver, augmenting HSC proliferation and sinusoidal vasoconstrictionviaincreased expression of Ang II type 1 receptor (AT1R) on HSCs[33-35]. Herath and colleagues[24]have shown that the upregulated AT1R in the cirrhotic rat liver responded to Ang II with an increased vasoconstrictive response compared to perfused control livers. As liver disease progresses, the activated HSCs augment the production of another potent vasoconstrictor peptide, endothelin-1 (ET-1). The HSCs become an autocrine target to ET-1 and actingviaendothelin receptors, ETRAand ETRB2, and increases HSC proliferation and contraction, thereby increasing hepatic vascular tone and resistance[36,37]. Moreover, cyclooxygenase -derived prostaglandins (prostaglandin H2) and thromboxane A2, and lipoxygenase-derived leukotrienes are also over-produced in the cirrhotic livers modulating hepatic vascular tone[38,39].
The elevated intrahepatic vascular tone is further affected by endothelial dysfunction which is characterised by a reduced intrahepatic bioavailability of vasodilators such as nitric oxide (NO), together with an increased release of vasoconstrictors[29,39,40]. Reduced activity of endothelial nitric oxide synthases (eNOS) is responsible for decreased hepatic NO production and impaired intrahepatic vasodilatation[41,42]. Reduced eNOS activity is mainly due to the binding of eNOS to the inhibitory protein caveolin-1 in venous and sinusoidal endothelial cells of the cirrhotic liver[43]. However, the contribution of many other factors has also been described, such as dysregulation of eNOS phosphorylation due to abnormal signaling by the regulator protein kinase B, reduced expression of eNOS activator G-protein coupled receptor kinase interactor-1, deficiency of eNOS co-factor tetrahydrobiopterin and activation of RhoA/Rho-kinase[44-47].
INCREASED SPLANCHNIC BLOOD FLOW
As outlined above, splanchnic vasodilation leading to increased portal blood flow also contributes to the pathogenesis of portal hypertension. In contrast to the presence of endothelial dysfunction to vasodilators in hepatic vasculature, in cirrhotic splanchnic vessels, vasodilatation is promoted by local over-production of vasodilators, which, along with intrinsic vascular hypocontractility allow increased blood flow through the splanchnic vessels.
Hyperactive splanchnic vascular endothelial cells over-produce NO in response to different stimuli such as shear stress, inflammatory cytokines and vascular endothelial growth factors (VEGF)[39,40,48,49]. The main isomer responsible for splanchnic vascular over-production of NO is eNOS. When released from the endothelial cells, NO diffuses into VSMCs, where it directly stimulates membrane-bound soluble guanylate cyclase to release cyclic guanosine monophosphate (cGMP) from guanosine triphosphate. Subsequently, cGMP induces K+efflux increasing intracellular Ca2+concentration and activates cGMP dependent protein kinase to dephosphorylate myosin light chain kinase, inducing vasodilatation[38,40](Figure 1). Vasodilatory molecules such as angiotensin-(1-7) [Ang-(1-7)][22], endogenous cannabinoids (ECs) and carbon monoxide (CO)[50]have been shown to regulate the vasodilatory pathwaysviaeNOS/NO dependent mechanisms.
Several lines of research evidence support the augmented role of eNOS in increased NO over-production in the splanchnic vascular bed in cirrhosis. It has been reported that there is an elevated eNOS activity and nitrite levels in portal circulation of human cirrhotic patients undergoing liver transplantation[51]. In agreement with this, elevated eNOS expression and activity has been reported in the splanchnic vessels of portal hypertensive rats[52-55]. A number ofin-vivoandin-vitrostudies have shown that eNOS inhibition with non-selective NOS inhibitor (iNOS), N(G)-nitro-L-arginine methyl ester, reduced vascular hypocontractility and plasma volume, and increased perfusion pressure in portal hypertensive rats[25,56-59], providing evidence for the importance of eNOS/NO system in splanchnic and systemic vasodilatation in cirrhosis. Supporting this, denudation of vessels to remove the endothelium ameliorated vascular hypocontractility, suggesting that over-production of NO is endothelium dependent[60,61].
However, NOS/NO is not the only system involved in altering splanchnic hemodynamics in cirrhosis. This fact is proven by the studies in which inhibition of NO or denudation of cirrhotic rat vessels failed to completely normalize the splanchnic vascular hypocontractility[56,58,62,63]. Moreover, eNOS and/or iNOS gene knockout mice still developed hyperdynamic circulation after the induction of portal hypertension[64]. This suggests that in addition to NO, other paracrine/autocrine vasodilators such as prostaglandin I2(PGI2), endothelium-derived hyperpolarizing factor(s) (EDHFs) including epoxyeicosatrienoic acids, hydrogen sulphide (H2S), CO, and ECs may contribute to the pathogenesis of hyperdynamic circulation and splanchnic vasodilatation in cirrhosis[40,65]. It is shown that molecules such as PGI2[66-68]and ECs[69]activate adenyl cyclase to increase cyclic adenosine monophosphate (cAMP) release in response to mechanical/humoral stimuli, thereby inducing the relaxation of VSMCs. The vasodilatory mechanism of H2S has been shown to be regulatedviaopening of K+-ATP channels[70]. In the absence of NO, EDHFs also act as prominent vasodilatory molecules, to induce vasodilatationviaarachidonic acid metabolism and activation of K+channels in the VSMCs[40,71,72]. Whilst both eNOS/NO system and EDHFs may participate in vasodilatory responses in the same vasculature, evidence also points to the possibility that the contribution of each system may depend on the size of the vessels[73]. This was based on the observations that different regions of rat superior mesenteric artery responded differently to acetylcholine-induced endotheliumdependent relaxation, and in comparison with large conduit vessels such as the aorta, it has been shown that the contribution of NO was most prominent in the aorta with highest expression of eNOS, whereas that of EDHF was most prominent in the distal mesenteric arteries accompanying the least eNOS expression[73-75].
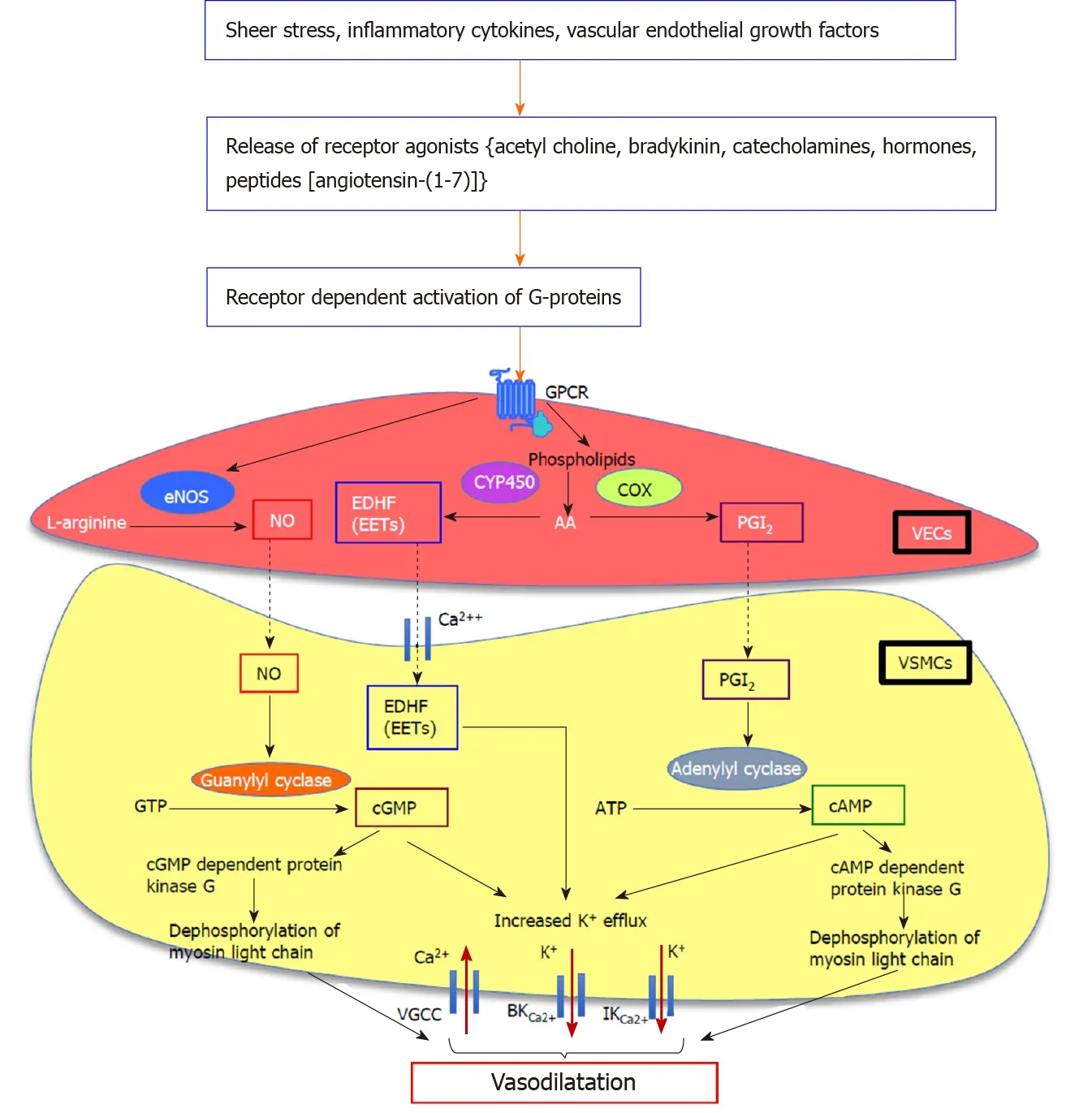
Figure 1 Proposed downstream signalling pathways of endothelium-dependent vasodilatation in splanchnic vasculature. Nitric oxide (NO) is a key mediator of mesenteric vasodilatation in cirrhosis. G protein-coupled receptor-mediated activation of G-proteins augments the production and activation of a number of vasodilatory pathways including endothelial nitric oxide synthase/nitric oxide (eNOS/NO), vasodilating prostacyclins, and metabolites of arachidonic acid (AA) metabolism which act as endothelium-derived hyperpolarizing factors such as epoxyeicosatrienoic acids (EETs) in vascular endothelial cells. The eNOS converts L-arginine to produce NO which diffuses into the vascular smooth muscle cells (VSMCs). In VSMCs, NO which activates membrane-bound guanylyl cyclase enzyme to release cyclic guanosine monophosphate from guanosine triphosphate, mediates vasodilatation by increasing K+ efflux via large-conductance and intermediate-conductance calcium-activated potassium channels and by decreasing Ca2+ influx via voltage-gated calcium channels, and by dephosphorylation of myosin light chain. In addition to NO, vasodilators such as prostaglandin I2, derived from AA by the action of cyclooxygenase enzyme which activates membranebound adenylyl cyclase enzyme to release cyclic adenosine monophosphate from adenosine triphosphate, causes vasodilatation by increasing K+ efflux and dephosphorylation of myosin light chain. The EETs deriving from AA by the action of endothelial epoxygenases such as cytochrome P450 directly act on Ca2+ and K+ channels, causing hyperpolarization of VSMC membrane and subsequent vasodilatation. NO: Nitric oxide; GPCR: G protein-coupled receptor; eNOS/NO: Endothelial nitric oxide synthase/nitric oxide; AA: Arachidonic acid; EDHFs: Endothelium-derived hyperpolarizing factors; EETs: Epoxyeicosatrienoic acids; VECs: Vascular endothelial cells; VSMCs: Vascular smooth muscle cells; cGMP: Cyclic guanosine monophosphate; GTP: Guanosine triphosphate; BKCa2+: Large-conductance; IKCa2+: Intermediate-conductance; VGCCs: Voltage-gated calcium channels; PGI2: Prostaglandin I2; COX: Cyclooxygenase; cAMP: Cyclic adenosine monophosphate; ATP: Adenosine triphosphate; CYP450: Cytochrome P450.
It should be noted that this vasodilated hyperdynamic circulatory state of cirrhosis persists despite the activation of powerful vasoconstrictor systems in the systemic circulation such as the RAS, endothelin system and sympathetic tone. It appears that this resistance to the vasoconstrictor agents is likely due to the presence of an intrinsic vascular hypo-responsiveness of the splanchnic/systemic vessels[28,30,76]. A number of human and animal studies have demonstrated a splanchnic vascular hyporesponsiveness to various vasoconstrictors including ET-1, Ang II, adrenoceptor agonists, neuropeptide Y and vasopressin in cirrhosis. Supporting this, a reduced level of portal ET-1 was found to be accompanied by an increased expression of the vasodilatory ETBreceptor in both mesenteric vascular endothelial and smooth muscle cells of cirrhotic rats[77]. A marked increase in norepinephrine concentrations has been detected in portal circulation of cirrhotic humans and rats, however it is proposed that the sustained sympathetic over-activation with norepinephrine may lead to vascular desensitization, thereby aggravating splanchnic vasodilatation in cirrhosis[78,79]. Similarly, Ang II plasma concentration is also elevated in cirrhosis[80], however it is speculated that the binding of receptor desensitizing proteins such as arrestin-2 and GRK2 to the AT1R may be responsible for vascular hyporeactivity to Ang II[81]. It has also been suggested that despite the attenuated reactivity to vasoconstrictors, expression and affinity of some receptors to their endogenous vasoconstrictors are not altered, and therefore, alterations in downstream signal transduction pathways is likely to be primarily responsible for this vascular hypo-reactivity[81-83]. However, this phenomenon of hypo-contractility of cirrhotic splanchnic vessels was questioned by a recent study published by our laboratory, demonstrating that pressor response of splanchnic vessels obtained from liver transplant recipients to Ang II was similar to that of control splanchnic vessels[84]. Taken together, these results imply more work is needed to clarify splanchnic vascular hypo-responsiveness to various vasoconstrictors in portal hypertension.
FORMATION OF PORTOSYSTEMIC COLLATERALS
The development of a venous vascular network of porto-systemic collaterals also accompanies the development of portal hypertension in cirrhosis. Formation of collaterals by activation of angiogenesis is modulated by several growth factors such as VEGF, placental growth factor, pigment epithelial derived factor and platelet derived growth factor (PDGF)[29,38,49,85]. Blocking VEGF and PDGF receptors with sorafenib administration for one month has been shown to reduce the extent of collateralization and portal pressure in cirrhotic rats[86,87], and reduced portal venous flow in patients with hepatocellular carcinoma[88]. Some studies also suggested that upregulated NO in splanchnic vasculature may also play a role in collateral formation[48]. Nicotinamide adenine dinucleotide phosphate oxidase has also been shown to play an important role in portal hypertension by modulating splanchnic angiogenesis and the formation of portosystemic collaterals[89].
CLINICAL DIAGNOSIS OF PORTAL HYPERTENSION
Portal hypertension is defined as a sustained increase in the pressure gradient between portal and systemic circulation. The most common parameter used to measure portal pressure is the hepatic venous pressure gradient (HVPG), the pressure gradient between the portal vein (PV) and inferior vena cava (IVC)[12]. In healthy individuals, the normal HVPG is 1-5 mmHg. In compensated cirrhosis, a HVPG between 6-10 mmHg portends mild subclinical portal hypertension, whilst clinically significant complications of portal hypertension develop when it exceeds 10 mmHg. Decompensated cirrhosis is characterised by an HVPG above 12 mmHg, which is manifested by the development of porto-systemic shunting, variceal rupture and bleeding[90,91]. Moreover, it has been reported that the probability of survival in cirrhotic patients with HVPG above 16 mmHg was below 70% of those patients who have an HVPG below this level and that this poor survival rate was related to Child-Pugh class[92]. The HVPG is a quantitative assessment but there are other indicators of PHT including the presence of varices on endoscopy, physical exam findings of splenomegaly, ascites and caput medusa as well as classical imaging findings on ultrasound, computer tomography and magnetic resonance imaging studies.
Portal pressure can be measured in experimental animals by direct cannulation of the PV or its tributary, the superior mesenteric vein. However, this is not feasible in the clinical setting and therefore, HVPG measurement has been adopted as the most common method for the monitoring of portal pressure. In this technique, a French balloon tipped catheter is inserted into the right internal jugular vein and advanced through right atrium and IVC into the hepatic vein (HV) under fluoroscopic guidance. Free hepatic vein pressure (HVP) is measured when the tip of the catheter is free in the HV. The balloon is then inflated occluding flow and creating a proximal static column of blood, which transmits the pressure from the hepatic sinusoids to the catheter. To obtain accurate and reliable pressure measurements, the readings are permanently traced and at least 3 pressure readings are obtained after HV occlusion to calculate the wedged hepatic venous pressure (WHVP)[93]. HVPG is the difference between WHVP and the HVP, and is a measure of sinusoidal pressure which is closely correlated with portal pressure[94]. In the normal liver, the HVPG is slightly lower than portal pressure, owing to pressure equilibrium through the interconnected sinusoids. However, in the cirrhotic liver the static column of blood created by occluding the hepatic vein cannot be decompressed at the sinusoidal level due to the disrupted connections between sinusoids, therefore, the HVPG gives a good approximation of the portal pressure in cirrhosis[90,93,94]. However, it should be noted that HVPG is a measure of sinusoidal pressure and does not accurately assess pre-sinusoidal portal hypertension that occurs in pre-cirrhotic primary biliary cholangitis.
ARTIFICIAL INTELLIGENCE IN PREDICTING CLINICALLY SIGNIFICANT PORTAL HYPERTENSION
Although HVPG is considered to be the gold standard in the measurement of portal pressure, this technique is an invasive and its reliability depends on the experience of the physician performing the procedure. Therefore, methods to evaluate the severity of liver fibrosis and stiffness such as liver transient elastography, ultrasound elastography, doppler and contrast-enhanced ultrasonography and serum tests, have been developed and validated as tools for the prediction of the presence of clinically significant portal hypertension and gastroesophageal varices using the assessment of cirrhosis extent as a predictor of the extent of portal hypertension[95,96].
Recent studies have focused on integrating artificial intelligence in the diagnosis of liver disease and portal hypertension. Such studies use computer-based programs to determine whether there is a correlation between different diagnostic markers and clinical outcomes. These computer programs use non-linear statistical analyses in order to establish a relationship between different input and output variables. A recent study using artificial neural networks (ANNs) compared the diagnostic performance of seven input variables (transient elastography and six serum tests) in predicting the presence of cirrhosis, clinically significant portal hypertension and oesophageal varices in patients[95]. This study revealed that liver stiffness measured by transient elastography could be used to detect portal hypertension and varices with > 80% accuracyviaANNs. Another study showed the effectiveness of a data mining software in establishing the relationships between various input variables, including clinical parameters, serum markers, liver ultrasonography and transient elastography, compared with portal pressure measured by HVPG[97]. This software used different classification meta-algorithms on selected datasets to show that there is a relationship between the above input variables and measured portal hypertension, suggesting this meta-algorithm analysis could be used as a substitute for the measurement of portal pressure by HVPG. Whilst these methods work well in advanced cirrhosis with established portal hypertension, they perform less well in earlier stages of liver disease.
CLINICAL MANIFESTATIONS OF PORTAL HYPERTENSION
Most events that occur during the evolution of chronic liver disease are the consequences of portal hypertension. Clinical manifestations of decompensated cirrhosis including the formation of varices and variceal bleeding, ascites, HRS and HPS have been directly related to the development of a hyperdynamic circulation, splanchnic vasodilatation and portal hypertension in cirrhosis. It should be noted that in 2016, HRS types 1 and 2 were reclassified as HRS-AKI and HRS-NAKI respectively, with the difference between the two dependent upon the trajectory of the creatinine rise above baseline[98,99]. The development of decompensation events is associated with a reduction in the median survival of a patient to less than 2 years, from 12 years in a cirrhotic patient without these complications[91].
The hyperdynamic circulation in cirrhosis is characterized by systemic and splanchnic vasodilatation, plasma volume expansion and increased cardiac index. Splanchnic vasodilatation results in an excessive blood volume in the splanchnic vascular compartment, leading to a reduction in effective arterial blood volume. Subsequently, endogenous neuro-hormonal systems such as the RAS and sympathetic nervous system are activated in an attempt to restore normal blood volumeviaincreasing sodium and water retention and by promoting vasoconstriction and increased cardiac output[11](Figure 2). However, in decompensated cirrhosis, these compensatory mechanisms are ineffective due to the intrinsic splanchnic vascular hypocontractility. Patients with advanced cirrhosis may also develop cirrhotic cardiomyopathy which limits the ability of the heart to increase cardiac output[26].
GASTROESOPHAGEAL VARICES
Development of gastroesophageal varices and associated massive upper gastrointestinal bleeding is the leading cause of death in cirrhotic patients. Varices are dilated collateral venous communications formed between portal and systemic circulations, mainly in the upper gastrointestinal region. Varices can divert 90% or more of the elevated portal flow back into heart. Eventually they remodel and progressively enlarge to accommodate increasing blood flow producing a risk of rupture and bleeding[12].
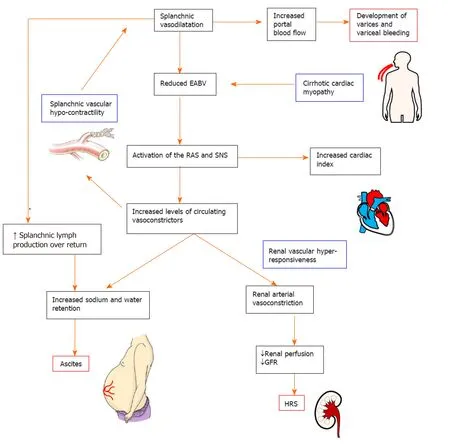
Figure 2 Pathophysiology of the complications in decompensated cirrhosis. Splanchnic vasodilatation and subsequent reduction of effective arterial blood volume (EABV) triggers the activation of homeostatic mechanisms such as the renin angiotensin system and sympathetic nervous system to promote sodium and water retention and vasoconstriction. Increased hydrostatic pressure and increased capillary permeability in splanchnic vessels cause leaking of excess fluid into peritoneal cavity and the onset of ascites. The renal vasculature is hyper-responsive to the activated circulating vasoconstrictor systems, creating a deficit in renal perfusion and glomerular filtration rate which in turn leads to the development of hepatorenal syndrome. In decompensated cirrhosis, the compensatory mechanisms to restore EABV are ineffective due to the intrinsic splanchnic vascular hypocontractility and cardiomyopathy. EABV: Effective arterial blood volume; RAS: Renin angiotensin system; SNS: Sympathetic nervous system; GFR: Glomerular filtration rate; HRS: Hepatorenal syndrome.
In cirrhotic patients, the collateral circulation begins to develop when HVPG rises above 10 mmHg. When HVPG is elevated above 12 mmHg, there is a significant risk of variceal rupture and bleeding[90,91]. By far the most clinically significant and relevant of these are the gastroesophageal varices, although they can also occur within stomach, rectum, omentum and elsewhere in the gastrointestinal tract. Gastroesophageal varices are more prone to bleed because of the lack of external tissue support and high trans-luminal pressure gradient due to negative oesophageal luminal pressure during inspiration[100,101].
Upon diagnosis of cirrhosis, varices are detected in about 30% and 60% of compensated and decompensated patients, respectively[102-104]. Patients without varices develop them at a rate of 8% per year. Long term follow-up studies revealed up to 90% of the cirrhotic patients will eventually develop varices. There is a 10%-30% risk for variceal rupture within the first year of diagnosis[105]. Despite the advances in the management of variceal bleeding in the past few decades, average 6-wk in-hospital mortality of first variceal bleeding still approaches 20%[100,106,107].
PHARMACOTHERAPIES TARGETING PORTAL HYPERTENSION IN CIRRHOSIS
Over the past few decades, a number of advances have been made in the development of novel treatment strategies to treat portal hypertension and its complications such as variceal bleeding. These therapies aim at reducing the risk of bleeding from varices by dropping the HVPG at least by 20% from baseline pressure or to less than 12 mmHg. However, the effectiveness of the currently available drugs including NSBBs in the treatment of variceal bleeding is limited, therefore there is an ongoing need for investigation and development of new therapies.
THERAPIES TARGETING SPLANCHNIC VASODILATATION
NSBBs
In 1981, the NSBBs were first introduced as a treatment for recurrent gastrointestinal bleeding in patients with cirrhosis[108]. Later, they were shown to be similarly effective in primary prophylaxis for variceal bleeding[109]. Since then, the effectiveness of NSBBs as first line pharmacotherapy in variceal bleeding has been documented by many clinical trials[110-114]. NSBBs lower HVPG by two principal mechanisms; firstly, by reducing cardiac output which leads to a subsequent reduction in splanchnic blood flow (cardiac 1-adrenergic blockade) and secondly by increasing splanchnic vascular resistance by allowing unopposed vasoconstrictive alpha-adrenergic activity thereby reducing portal blood flow (splanchnic 2-adrenergic blockade)[26,115-117].
American Association for the Study of Liver Disease guidelines, British Society of Gastroenterology guidelines and the Baveno VI consensus recommend the use of NSBBs in both primary and secondary prophylaxis of variceal bleeding[118-120], and their effectiveness has been demonstrated by a number of clinical trials. In patients with existing small varices, NSBBs slowed the progression of small varices into large varices (11% and 37%, NSBBvsplacebo control, respectively) and decreased the incidence of first variceal hemorrhage (12% and 22%, NSBBvsplacebo control, respectively)[121]. In patients with medium or large varices and at a high risk of bleeding, they reduced the risk of bleeding to 14% from 30% in control subjects[122]. The effectiveness of NSBBs in secondary prophylaxis has been shown by studies which demonstrated that at one year of treatment with propranolol, the drug reduced recurrent bleeding of cirrhotic patients with gastrointestinal bleeding[108,111]. However, NSBBs are not similarly effective in preventing the development of new varices in early cirrhotic patients with mild subclinical portal hypertension[117]. Moreover, these drugs are contraindicated in patients at the time of acute variceal bleeding due to their systemic hypotensive and cardiac side effects[123].
Despite their proven clinical effectiveness, traditional NSBBs such as propranolol and nadolol are contraindicated or poorly tolerated in up to 15%-20% of patients and up to 60% of patients do not achieve any therapeutically useful fall in HVPG with these NSBBs[105,124,125]. NSBBs produce a number of cardiac and non-cardiac adverse effects, such as headaches, fatigue, asthma and shortness of breath, which led to the discontinuation of treatment in about 15% of the patients in clinical trials[126,127].
The 1-adrenergic effects of NSBBs in reducing cardiac output has detrimental side effects in patients with advanced cirrhosis. In end stage cirrhotic patients with refractory ascites and/or SBP, NSBBs blunt the ability to maintain an adequate systemic arterial blood pressure, and defective renal perfusion may lead to the development of HRS[128,129], and in end stage cirrhosis NSBBs have been associated with increased mortality[130].The use of NSBBs is also contraindicated in around 15% of the cirrhotic patients who have asthma, chronic obstructive pulmonary diseases poor cardiac function or low blood pressure[91].
The traditional and most widely used NSBBs in clinical practice are propranolol and nadolol. Many clinical trials suggest that both drugs have a similar effectiveness in primary prophylaxis of variceal bleeding. However, some studies showed that the side effects produced by nadolol (9%-13%) were lower compared to those of propranolol (12%-31%), although direct comparison between the two groups has not been performed[131]. Moreover, compared to propranolol, nadolol has a longer half-life in the circulation attributed to its low lipid solubility and hepatic metabolism, thus patients can be dosed less frequently[132].
Similar to propranolol, the NSBB timolol is effective in reducing HVPG, particularly in early cirrhosis (12% and 13% reduction in timolol and propranolol, respectively)[133,134]. Other NSBBs used as antihypertensive drugs including sotalol, pindolol and penbutalol may also be effective in the treatment of portal hypertension; however, the efficacy of these drugs has not yet been tested in patients with oesophageal varices.
The NSBB, carvedilol, has been shown to be more effective than propranolol in reducing portal pressure[135]. In one clinical trial, carvedilol and propranolol resulted in a HVPG reduction of more than 20% from baseline values, or to less than 12 mmHg, in 64% and 14% of cirrhotic patients, respectively[136]. In addition to its potent -blocker activity on reducing splanchnic vasodilatation, the effectiveness of carvedilol is likely due to its potential anti-1 adrenergic activity, which is about one-tenth of its -blocker activity, thus reducing intrahepatic vascular resistance[137]. Thus, whilst carvedilol decreases cardiac output with concomitant increase in splanchnic resistance by blocking the receptors, its anti-1 receptor activity helps decrease hepatic vascular tone and hepatic resistance[138]. Moreover, carvedilol is also known to have antioxidant, antifibrotic and anti-inflammatory properties, which might potentially benefit patients with advanced cirrhosis[132]. However, the vasodilating anti-1 effect of carvedilol has potential to enhance systemic hypotension, leading to a reduction in renal perfusion and therefore, its clinical application is limited, especially in cirrhotic patients with refractory ascites and in patients with advanced, decompensated cirrhosis[136,139]. However, labetalol, another NSBB with similar anti-1 adrenergic activity failed to reduce HVPG in patients with cirrhosis[140]. Although individual studies demonstrated that carvedilol may be more effective than traditional NSBBs such as propranolol in preventing variceal bleeding in cirrhotic patients, a recent meta-analysis which systematically analysed ten randomized clinical trials using carvedilol, propranolol and nadolol, concluded that carvedilol is not as effective as traditional NSBBs in reducing mortality, variceal bleeding and serious adverse events[141]. Thus, the authors concluded that additional evidence is required from adequately powered, long-term, double-blind, randomised clinical trials, evaluating both clinical and haemodynamic outcomes. Importantly, in the United Kingdom, there are adequately powered randomized clinical trials in progress comparing the effectiveness of carvedilolvsplacebo[142]and carvedilolvsendoscopic variceal ligation therapy[143]in cirrhotic patients and the outcomes of these studies are yet to be published. These are the first trials investigating the effectiveness of carvedilol on variceal bleeding in cirrhotic patients.
Selective 1 receptor blockers such as atenolol and metoprolol also reduce HVPG in cirrhotic patients. The reduction of HVPG achieved with these selective blockers is related to their 1 receptor blockade-mediated effect on cardiac index, which contrasts with the cumulative cardiac (1) and splanchnic (2) effects of NSBBs such as propranolol. It was reported that the efficacy of atenolol in reducing HVPG and variceal bleeding was less and not sustained in cirrhotic patients compared to propranolol[144,145]. Similarly, treatment with metoprolol also was associated with higher rate of recurrent bleeding[146]. Based on these findings and also due to their profound cardiac side effects, selective 1 receptor blockers are not therefore recommended in the prophylaxis of variceal hemorrhage[125].
OTHER THERAPIES TARGETING SPLANCHNIC VASODILATATION
Splanchnic vasoconstrictors such as vasopressin, somatostatin and their analogues are used alone or in combination with endoscopic therapy to treat patients with acute variceal bleeding[118].
Similar to NSBBs, vasopressin reduces portal pressure by producing splanchnic vasoconstriction and thereby decreasing portal venous blood flow. However, due to its potent vasoconstrictive ability vasopressin has been shown to produce multiple side effects in the heart and systemic circulation including hypertension, ventricular arrhythmias, cardiac and peripheral ischemia, and bowel ischemia[131]. Combination therapy of vasodilator nitroglycerine has been shown to counteract the cardiac side effects of vasopressin in cirrhotic patients and experimental animals[147]. The synthetic vasopressin analogue terlipressin which is selective for the V1 vasopressin receptor has significantly fewer side effects and therefore, it is recommended for patients with acute variceal bleeding[148,149]. However, terlipressin is not available in some countries including the United States and Canada.
Treatment with somatostatin and its analogues such as octreotide and vapreotide is also recommended for the treatment of acute variceal haemorrhage[150]. Among these, only octreotide is readily available in many countries including the United States. Although these drugs were previously shown to promote splanchnic vasoconstrictionviainhibiting the release of vasodilatory peptide glucagon, it is now known that the local adrenergic 1 receptor-mediated vasoconstrictive effects of somatostatins are responsible for counteracting the splanchnic vasodilatation. A recent meta-analysis showed that the efficacy of somatostatin/octreotide on variceal re-bleeding was similar to that of vasopressin/terlipressin[151]. In current guidelines, however, they are recommended for acute variceal bleeding[152], although they may have side-effects such as tachyphylaxis, bradycardia and hypertension[153,154].
THERAPIES TARGETING INCREASED INTRAHEPATIC VASCULAR TONE
Reduction of intrahepatic and/or porto-collateral resistance using vasodilatory agents such as nitrates could also be an ideal therapeutic approach in the treatment of portal hypertension in cirrhosis. These agents such as isosorbide-5-mononitrate (ISMN), are ineffective when given as monotherapy but when given as a combined treatment with an NSSB improve the efficacy of NSBBs in the reduction of HVPG, and may be very useful in secondary prophylaxis for variceal bleeding[155]The combined treatment, has been shown to be effective for those patients who are non-responders to standard NSBB treatment alone[121,156].
Selective 1 adrenergic receptor blockers such as prazosin are another experimental therapy for the modification of intrahepatic vascular tone. A study in 12 patients with oesophageal varices demonstrated that short-term and long-term administration of prazosin reduced HVPG by 25% and 19% of the baseline, respectively[157]. However, prazosin produced long term deleterious side effects in cirrhotic patients by promoting peripheral vasodilatation and enhancing sodium and water retention, leading to the development of ascites. A study comparing the effects of combination therapy of propranolol with ISMN or prazosin showed that propranolol plus prazosin treatment reduced HVPG in cirrhotic patients more than propranolol plus ISMN treatment[158]. However, systemic hypotension was more evident with combination therapy of prazosin compared to ISMN.
Statins are another category of therapy that has been widely studied in the modulation of intrahepatic resistance and vascular tone. These drugs have been shown to selectively enhance eNOS activity in cirrhotic livers, promoting the production of NO in hepatic sinusoidal endothelial cells, thus they appear to behave as “true liverselective vasodilators” in cirrhosis[47,159,160]. In the cirrhotic liver, statins also reduce the contraction and proliferation of myofibroblastic HSCs by blocking RhoA-dependent non-canonical hedgehog pathway[161]. In addition, statins have been shown to reduce neoangiogenesis and collateral flow by blunting the non-canonical hedgehog signalling pathway in experimental cirrhosis[161].
A number of clinical studies have highlighted the potential importance of the use of statins such as simvastatin and atorvastatin in the treatment of portal hypertension in cirrhosis. Recently, a systematic review and meta-analysis summarized the results of 13 studies that used statins in patients with chronic liver disease, to show that these drugs significantly reduce the progression of chronic liver disease into decompensated cirrhosis by 46%, patient mortality by 46%, and the risk of variceal bleeding and progression of portal hypertension by 26% in cirrhotic patients[162]. An important multicenter double blind randomized controlled trial demonstrated that the treatment with simvastatin significantly reduced of HVPG (by 8.3%) in cirrhotic patients with portal hypertension, without inducing arterial hypotension, supporting the argument that these drugs are ‘true liver-selective vasodilators’[163]. Simvastatin also produced an additive effect on reducing HVPG in the patients receiving NSBB treatment (by 11%), possibly related to increased bioavailability of NO in the hepatic sinusoidal circulation[116]. In another recent clinical trial, addition of simvastatin to the NSBB treatment decreased overall mortality in patients although it failed to reduce the rate of re-bleeding from varices[164]. Whilst this study showed that 3% of the patients treated with simvastatin developed severe adverse side effects such as rhabdomyolysis, a more recent clinical trial suggested that statins-associated risks appear to be doserelated[165]. The above clinical evidence suggests that statins possess a clear potential for the treatment of portal hypertension in cirrhotic patients, however, further randomized clinical trials are warranted involving larger patient populations with clear clinical end points.
A recent study in experimental animal models suggests that statins may have divergent effects in cirrhotic and non-cirrhotic portal hypertension. Uschner and colleagues[161]showed that in a non-cirrhotic portal hypertensive rat model, statins worsened the portosystemic shunt flow and portal hypertension,viaincreasing extrahepatic angiogenesis through the canonical hedgehog pathway, suggesting that in a clinical setting, the use of statins could possibly be limited to cirrhotic patients with portal hypertension.
Selective hepatic delivery of vasodilators such as NO has also been identified as an ideal strategy in the modification of intrahepatic vascular tone. Unexpectedly, delivery of liver specific NO donor NCX-100 failed to decrease HVPG in a phase II clinical trial[166]. In addition, antioxidants such as ascorbic acid, caffeine and dark chocolate are proposed as therapies that improve endothelial dysfunction in cirrhosis[167-169]. Experimental evidence also suggests that multikinase inhibitors such as sorafenib could be used to inhibit collateral formation and/or neoangiogenesis by blocking both VEGF and PDGF receptors. However, the challenge with this therapy is the inability of the drug to differentiate pathological neoangiogenesis from physiological neoangiogenesis such as the cyclic changes of female reproductive tract[86,87,170].
The selective 2 adrenergic receptor agonist clonidine is another potential therapy for the treatment of portal hypertension in cirrhosis. 2 receptor agonists diminish sympathetic activity and reduce cardiac output and increase portal venous tone, thus they are expected to reduce portal venous inflow. Studies have shown that clonidine reduced HVPG by > 10% from baseline in around 87% of alcoholic cirrhotic patients[171]. However, the usefulness of clonidine as a treatment for portal hypertension is limited by deleterious systemic and renal side effects[172].
THE RAS
The RAS has long been recognized as a simple enzymatic cascade responsible for the regulation of cardiovascular and fluid homeostasis. However, understanding of this system has dramatically changed over the last few decades. It is now known that besides its physiological role, the RAS also mediates complex regulatory functions involved in disease progression and wound healing[173,174].
The RAS is comprised of two axes that function interdependently (Figure 3). The “classical axis” of the RAS comprises angiotensin converting enzyme (ACE), the vasoconstrictive peptide Ang II and its receptor AT1R. ACE cleaves the biologically inactive decapeptide angiotensin I (Ang I) to the octapeptide Ang II. Ang II is a potent vasoconstrictor that actsviathe AT1R in regulating blood pressure, and fluid and electrolyte homeostasis[53,175]. In addition to its vasoactive role, Ang II plays an important roles in the wound healing responsesviacell proliferation, tissue inflammation and fibrogenesis[176-180].
It is now known there is an “alternate axis” of the RAS comprising its active carboxypeptidase of angiotensin converting enzyme 2 (ACE2, a structural homologue of ACE), the effector heptapeptide Ang-(1-7) and its receptor Mas. ACE2 cleaves a single C-terminal residue from Ang II and Ang I to produce Ang-(1-7) and Ang-(1-9), respectively[181-183]. In comparison with ACE, the affinity of ACE2 for cleaving Ang I is low; however, ACE2 has more than 400-fold greater affinity to cleave Ang II to produce Ang-(1-7)[24,184-187]. Ang-(1-7) can also be formed from Ang I and Ang-(1-9) by the enzymatic action of neutral endopeptidase neprilysin and ACE, respectively[24,187,188].
In 1988, Schiavone and colleagues[189]published the first report of a biological action of Ang-(1-7) characterizedin vitro, which was immediately followed by a report showing anin vivoaction of the peptide[190]. Following these discoveries, a large body of research proved that it is an active peptide which has opposing effects to Ang II, including vasodilatation[25,191-194]. In addition, Ang-(1-7) also has anti-inflammatory, anti-fibrotic and anti-proliferative properties, thus attenuating disease progression, including chronic liver[80,195]cardiac[196-199]and renal[200-204]diseases. Available evidence suggests that the vasodilatory effects of Ang-(1-7) are regulated through increased production of NO[205-209]viaactivation of eNOS in endothelial cells[25,209,210]. It has been shown that Ang-(1-7) regulates Ser1177/Thr495 phosphorylation to increase eNOS activity and NO productionviaa P13K/Akt sensitive pathway[209]. It has also been shown that Ang-(1-7) mediates vasodilatation by potentiating bradykinin, the effector peptide of the kallikrein-kinin system, to release NO and vasodilatory prostacyclins. This suggests that kallikrein-kinin system and eNOS both contribute to Ang-(1-7)-mediated vasodilatation[205,208,211-213].
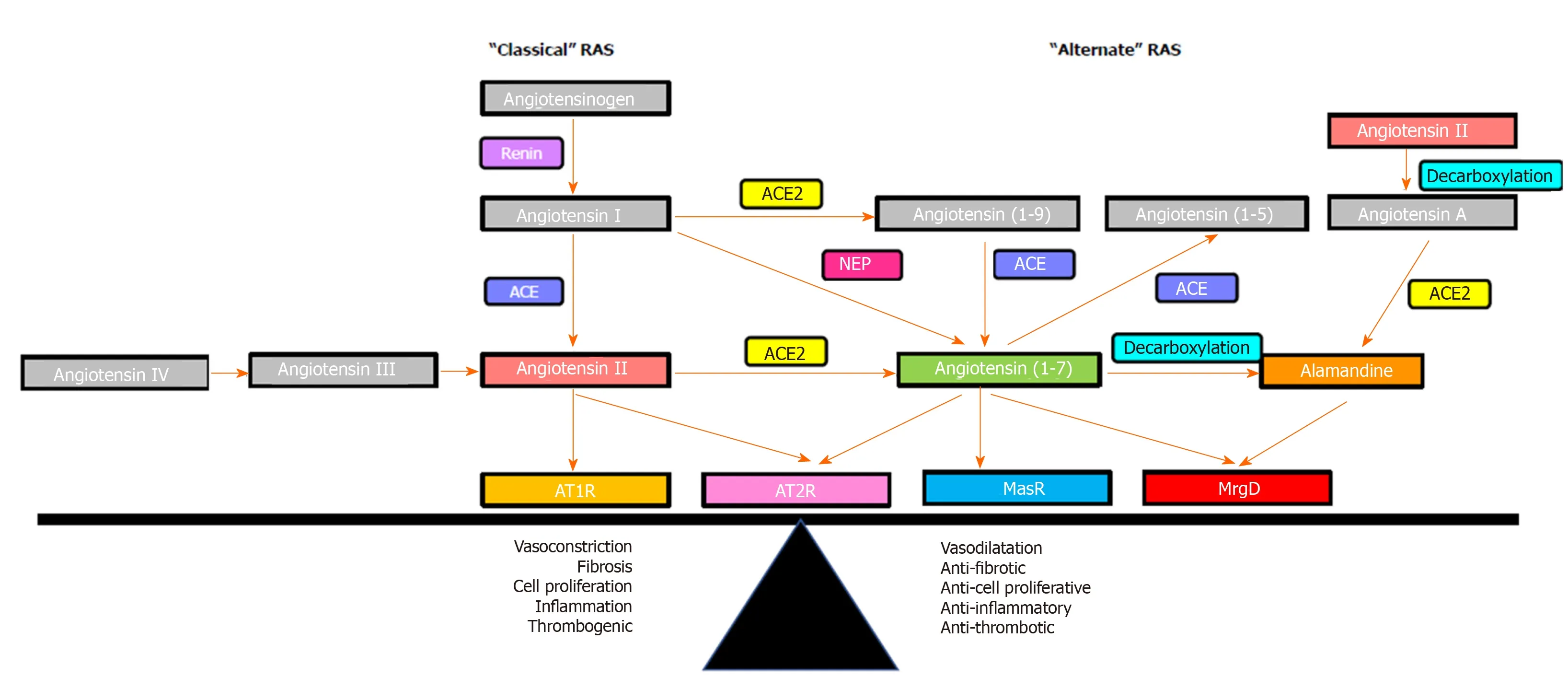
Figure 3 Overview of the renin angiotensin system. The effects of the renin angiotensin system (RAS) are determined by the balance between its “classical axis” and the “alternate axis”. Classical axis comprises of angiotensin converting enzyme, angiotensin II, angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R), which mediate vasoconstrictive (AT1R) or vasodilatory (AT2R) functions, proinflammatory and profibrogenic pathways. The alternate axis comprises of angiotensin converting enzyme 2, angiotensin-(1-7) and the Mas receptor opposes the effects of the classical RAS[53]. Recent studies identified that the vasodilatory action of angiotensin-(1-7) is also mediated via the Mas-related G protein-coupled receptor-type D, which also mediates the action of alamandine. RAS: Renin angiotensin system; ACE: Angiotensin converting enzyme; ACE2: Angiotensin converting enzyme 2; AT1R: Angiotensin II type 1 receptor; AT2R: Angiotensin II type 2 receptor; MasR: Mas receptor; MrgD: Mas-related G protein-coupled receptor-type D.
Identification of Ang-(1-7) as a functional peptide of the alternate RAS provided impetus to further investigate its mechanism of action. Evidence for the presence of a specific Ang-(1-7) receptor came in a study which showed thatin vivoandin vitrofunctional responses to Ang (1-7) were abolished by the synthetic Ang-(1-7) analogue D-Ala7-Ang-(1-7) (A779), whereas the effect of Ang-II could not be blocked by A779[214]. Subsequently, radioligand binding studies in bovine aortic endothelial cells demonstrated that both A779, and a non-peptide Ang-(1-7) agonist AVE0991 competed for binding with Ang-(1-7), which led to the speculation that Ang-(1-7) should have a specific receptor[215,216]. In 2003, Santos and colleagues[217]discovered the functional receptor for Ang-(1-7) and named it as the Mas receptor (MasR), which is a G protein-coupled receptor encoded by the Mas protooncogene. Specificity of MasR to Ang-(1-7) was demonstrated by Ang-(1-7) failing to bind to the kidneys of MasR knockout mice (MasR-KO), and Ang-(1-7) failing to exert a vasodilatory response in aortic rings isolated from these mice. Moreover, mesenteric arterial vessels isolated from MasR-KO mice showed a blunted vasodilatory response to acetylcholine, suggesting an endothelial dysfunction in Mas-KO mice, which confirmed the role of MasR in regulating the vascular tone[218,219]. Confirming the role of MasR in modulating vascular tone, Xu and colleagues[220]have shown that MasR deletion results in increased blood pressure and endothelial dysfunction. In other studies, MasR deletion altered ventricular function, vascular resistance and increased the profibrotic profile and Ca2+regulation in cardiomyocytes, further implying the functional significance of the Ang-(1-7)/MasR axis[221,222].
However, more recent studies have speculated on the existence of a second receptor for Ang-(1-7) which appears to play a leading role in controlling vascular tone. Initial evidence for this came from a study which showed that vasodilatory effects of Ang-(1-7) in rat aorta could not be blocked by MasR blocker A779, but were completely abolished by a synthetic Ang-(1-7) antagonist D-Pro7-Ang-(1-7) (D-Pro)[223]. Subsequently, work from our laboratory showed that in the cirrhotic perfused rat liver, the perfusion pressure reduction effect of Ang-(1-7) was blocked by D-Pro but not by A779, suggesting the existence of a receptor subpopulation other than MasR that is sensitive to D-Pro[25]. Lautner and colleagues[224]were able to characterize a novel receptor, which they identified as the Mas-related G protein-coupled receptor type-D (MrgD), that is activated upon binding to the newly discovered vasodilatory RAS peptide alamandine. Importantly, they found that the vasodilatory effects of alamandine were blocked by D-Pro, but not by A779. This confirmed previous reports that COS cells over-expressed with either MasR or MrgD released arachidonic acid in response to Ang-(1-7) stimulation, suggesting that both the MasR and MrgD mediate Ang-(1-7) action by releasing arachidonic acid, a precursor molecule of prominent intracellular vasodilating signaling pathways[225](Figure 1). More recently, another study strongly supported the concept that MrgD is a second receptor for Ang-(1-7) by showing that in the absence of either MasR or MrgD activation, mesangial cells failed to increase cAMP release in response to Ang-(1-7), and both A779 and D-Pro abolished cAMP release by HEK293 cells transfected with MasR or MrgD[226]. This study also showed that MrgD-KO mice had an impaired hemodynamic response to Ang-(1-7) administration. These findings collectively confirmed that MrgD is a functional receptor mediating the vasodilatory effects of Ang-(1-7).
THE RAS IN PORTAL HYPERTENSION
It is well known that the RAS is activated in cirrhosis and plays a major role in the development of portal hypertension[4,173,227,228](Figure 4). In cirrhotic patients and animal models of cirrhosis, the components of both classical and alternate axes of the RAS are markedly upregulated[34,53,80]. Work from our laboratory and others showed that the components of the classical RAS, in particular ACE and Ang II are significantly elevated in the circulation and the liver of cirrhotic animals, contributing to liver fibrosis and increased intrahepatic vascular tone[25,35,229-231]. Consistent with this, blockade of the classical RAS has been shown to improve experimental liver fibrosis and intrahepatic vascular resistance[24,229,232-234]. Recent discoveries demonstrate that the components of the alternate RAS including ACE2 and Ang-(1-7) are also upregulated in the circulation and the liver of cirrhotic animals[35,80,230]. Importantly, these RAS components are also increased in the splanchnic vascular bed of cirrhotic animals and have been suggested to play a pivotal role in splanchnic vasodilatation and the development of portal hypertension[22,23]. Therefore, manipulation of the RAS, particularly the alternate RAS, may provide a novel approach to treat portal hypertension in cirrhosis.
THE RAS AND INTRAHEPATIC VASCULAR RESISTANCE
In cirrhotic livers, Ang II levels are markedly increased, promoting HSC proliferation and ECM production[19,235], which increases intrahepatic resistance to the portal flow. Inhibition of Ang II actionviaACE inhibitors (ACEi) and/or angiotensin receptor blockers (AT1R blockers) (ARBs), has therefore been shown not only to attenuate hepatic fibrosis but also to alleviate intrahepatic resistance in animal models of cirrhosis[229,236,237].
In addition, the activated contractile phenotype of HSCs express increased AT1R and contract in response to Ang II and thereby increase sinusoidal vasoconstriction, leading to increased intrahepatic vascular resistance[33,175]. Supporting this phenomenon, work from our laboratory showed that the vasoconstrictive response to Ang II administration is increased in the perfused cirrhotic rat liver preparation compared to that in healthy liver, presumablyviathe effects of Ang II on upregulated AT1R in VSMCs cells and sinusoidal myofibroblastic HSCs[24]. This study also demonstrated that hepatic production of Ang II is increased in cirrhosis, suggesting that locally produced Ang II contributes to increased intrahepatic vascular resistance and highlighting the importance of the hepatic RAS in the regulation of portal hypertension.
It has been suggested that increased local expression of the components of the alternate RAS may be effective in counteracting the detrimental effects of the classical RAS in the cirrhotic liver[53]. Supporting this concept, work published by our laboratory demonstrated that liver-specific sustained over-expression of ACE2 ameliorated liver fibrosis and improved hepatic perfusion by reducing hepatic levels of profibrotic and vasoconstrictor peptide Ang II with concomitant increase in antifibrotic and vasodilator peptide Ang-(1-7)[238]. These findings supported earlier work published by our laboratory that infusion of Ang-(1-7) into cirrhotic rats significantly improved hepatic fibrosis by suppressing HSC activation, although portal pressure was not measured in this study[80]. Direct support for a vasodilatory action of Ang-(1-7) in the cirrhotic liver comes from work performed in our laboratory which showed thatin situperfused cirrhotic rat liver preparations pre-constricted with Ang II or methoxamine underwent a marked endothelium-dependent, and AT1R/AT2R independent, vasodilatory response to the infusion of Ang-(1-7)[25]. Moreover, work by our laboratory also demonstrated that administration of Ang-(1-7) or the non-peptide MasR agonist AVE0991 reduced the activation of primary rat HSCs in culture whereas the MasR blocker A779 increased their activation, as reflected by increased -smooth muscle actin expression. In other words, the antifibrotic and vasodilatory effects that can be produced by components of the alternate RAS would be expected to improve portal hypertension by reducing intrahepatic vascular resistance.
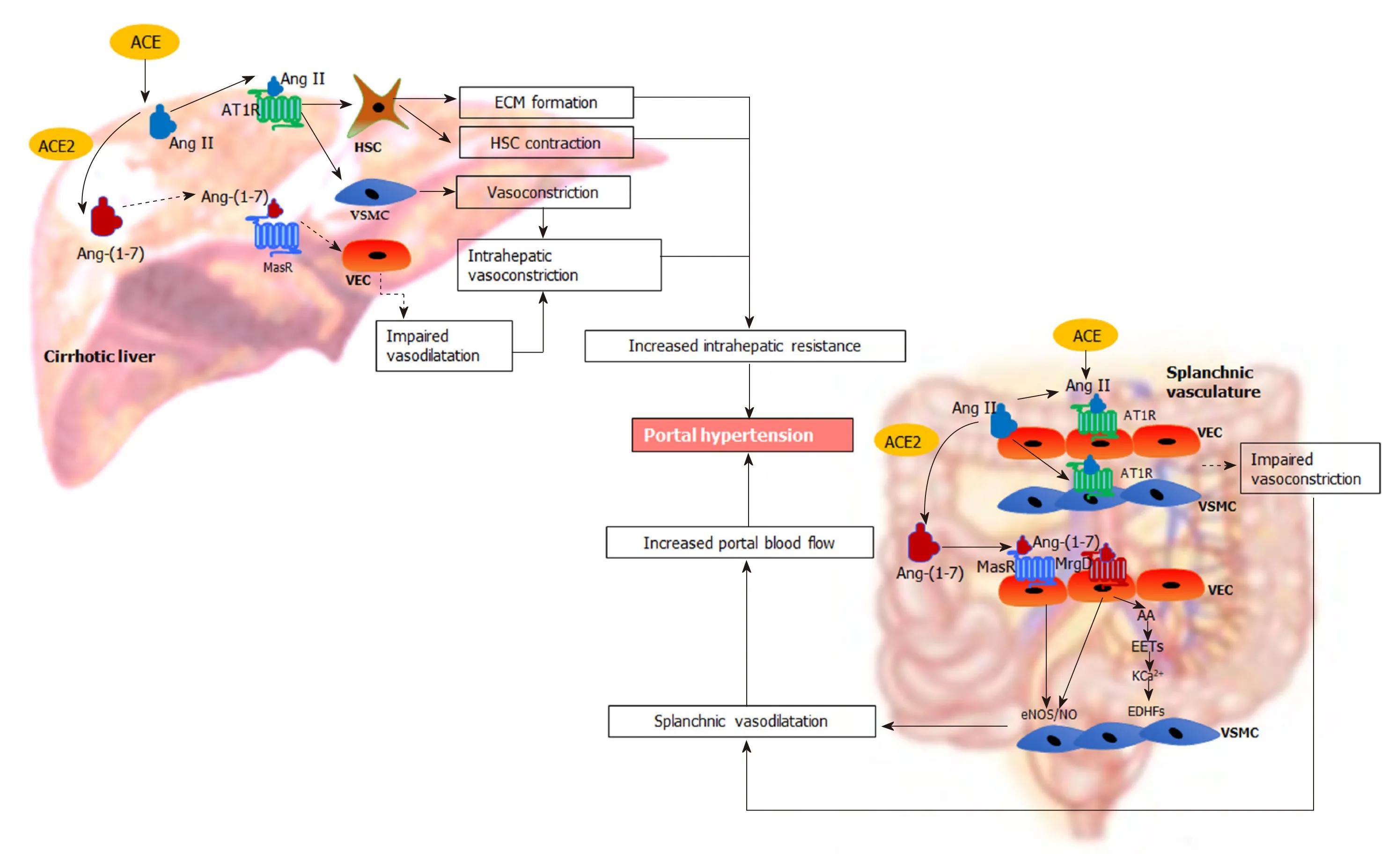
Figure 4 Renin angiotensin system-mediated pathophysiological changes in portal hypertension.In the cirrhotic liver, the “classical axis” of the renin angiotensin system (RAS) predominates. The vasoconstrictor octapeptide angiotensin II (Ang II), via the Ang II type 1 receptor in hepatic stellate cells (HSC) increases the deposition of extracellular matrix proteins, creating a fixed barrier to portal blood flow within the cirrhotic liver. In addition, Ang II also increases intrahepatic vascular tone by stimulating contraction of activated HSCs and vascular smooth muscle cells (VSMCs). Reduced release of vasodilating intracellular signalling molecules such as nitric oxide (NO) from vascular endothelial cells (VECs) by the action of endothelial NO synthase further impairs intrahepatic vasodilatation. In contrast, the “alternate axis” of the RAS predominates in the cirrhotic splanchnic vascular bed. The vasodilator heptapeptide angiotensin-(1-7), via its Mas receptor and, the Mas related G protein coupled receptor type-D, increases the release of NO from VECs to promote the relaxation of VSMCs causing splanchnic vasodilatation. This condition may be exacerbated by intrinsic vascular hypocontractility of mesenteric arteries to vasoconstrictors such as Ang II. Other than NO activity, release of other potent endothelium-derived hyperpolarizing factors such as epoxyeicosatrienoic acids derived from arachidonic acid of membrane phospholipids may play an important role in splanchnic vasodilatation by increasing K+ efflux through Ca2+-activated K+ channels in VSMCs. These lead to an increased portal blood flow, resulting in increased portal pressure. ACE: Angiotensin converting enzyme; ACE2: Angiotensin converting enzyme 2; Ang II: Angiotensin II; AT1R: Angiotensin II type 1 receptor; Ang-(1-7): Angiotensin-(1-7); MasR: Mas receptor; MrgD: Mas-related G protein-coupled receptor-type D; HSC: Hepatic stellate cells; VSMCs: Vascular smooth muscle cells; VECs: Vascular endothelial cells; ECM: Extracellular matrix; eNOS/NO: Endothelial nitric oxide synthase/nitric oxide; EDHFs: Endothelium-derived hyperpolarizing factors; EETs: Epoxyeicosatrienoic acids; AA: Arachidonic acid; KCa2+: Ca2+-activated K+ channels.
THE RAS AND SPLANCHNIC VASCULAR RESISTANCE
In contrast to the effect of vasoactive molecules in the intrahepatic vasculature, the systemic and splanchnic vessels are hyporesponsive to circulatory vasoconstrictors including Ang II and thus, these vessels remain dilated in cirrhosis[239-241].
ACE2 and Ang-(1-7) production are increased in the splanchnic vasculature of cirrhotic rats and cirrhotic patients compared to healthy controls, suggesting a potential role of Ang-(1-7) in splanchnic vasodilatation in cirrhosis[22]. Interestingly, in the systemic circulation Ang-(1-7) levels are increased with the progression of liver disease but Ang II rises only after the establishment of portal hypertension[35,80,242]. In cirrhotic patients at liver transplant, the Ang-(1-7)/Ang II ratio is elevated in the splanchnic circulation compared to systemic circulation, and is negatively correlated with the systemic vascular resistance, suggesting that there is an augmented activity of local Ang-(1-7) in the splanchnic vascular compartment[242]. Functional evidence for this association comes from work published by our laboratory that infusion of Ang-(1-7) reduced splanchnic vascular resistance in cirrhotic rats to a much greater degree than in controls, suggesting that Ang-(1-7) is a key mediator of splanchnic vasodilatation in cirrhosis[22]. Consistent with enhanced Ang-(1-7) activity, MasR is also upregulated in both human and rat cirrhotic splanchnic vessels, and the MasR blocker A779 blocked the vasodilatory effects of Ang-(1-7) and greatly reduced splanchnic vasodilation in cirrhotic ratsin vivo[22]. Moreover, we have recently demonstrated for the first time that in addition to MasR, the alternate receptor for Ang-(1-7), MrgD, is also upregulated in the splanchnic vessels of the cirrhotic rats[23]. This study demonstrated that both MasR and MrgD blockade improved portal hypertension, likelyviathe inhibition of Ang-(1-7) mediated splanchnic vasodilatation[23].
Dilatation of the splanchnic vascular bed has also been linked to intrinsic vascular hyporesponsive to vasoconstrictors including Ang II in cirrhosis[243-245]. Supporting thisin vitrovessel work, vasoconstriction measured by evaluating forearm blood flow responses to intra-arterial administration of Ang II was found to be lower in cirrhotic patients compared to the healthy controls[246]. Mechanistic insights into this was provided by studies which demonstrated that AT1R expression was either normal or upregulated in splanchnic vessels from cirrhotic patients[81,83], suggesting that vascular hyporesponsive to vasopressors is likely due to the changes that occur downstream of AT1R[82,247]. In support of this phenomenon, Ferlitsch and colleagues[240]demonstrated that a reduced response of forearm blood flow to intra-arterial administration of vasopressors including Ang II in cirrhotic patients was completely restored to that in control subjects by coadministration with vitamin C, suggesting that generation of intracellular reactive oxygen species, which is closely linked to the activation of AT1R, may be responsible for hyporesponsive to Ang II. These findings were however not in agreement with earlier findings which showed that AT1R-independent G-protein stimulation with AlCl3/NaF induced an intact contractile response of hepatic artery which is similar in magnitude to that of the control vessels obtained from organ donors and suggested that hyporesponsive to Ang II may be related to a receptorspecific phenomenon localized upstream from the G-protein level[243]. In marked contrast to these findings however, recent work published by our laboratory using splanchnic arterial vessels isolated from liver transplant recipients demonstrated that they were not hyporesponsive to the pressor effect of Ang II[84]. Thus, there is conflicting literature data regarding the vasoactivity of splanchnic arteries[84,243,245]and forearm arteries[240,246]to vasopressors such as Ang II, implying more work is needed to clarify the vasoactivity of mesenteric arterial vessels in portal hypertension as not only different vascular beds may respond differently to vasopressors but also differences in vasoactive responses may be due to the compounding effects of different experimental conditions[248].
MANIPULATION OF THE RAS IN PORTAL HYPERTENSION
It is clear that the RAS contributes to the pathogenesis of portal hypertension. Whilst inhibition of classical RAS is expected to reduce intrahepatic vascular tone, inhibition of the alternate RAS in splanchnic vasculature would be expected to improve portal hypertension by increasing splanchnic vascular resistance. Thus, both the classical and the alternate RAS are potential targets that can be manipulated in portal hypertension.
TARGETING THE CLASSICAL RAS IN PORTAL HYPERTENSION
Since the primary contribution of classical RAS to increased intrahepatic resistance in cirrhosis was identified through the evidence from studies in experimental cirrhosis, the effect of inhibition of the classical RAS has been studied in a number of clinical trials. The anti-portal hypertensive effect of ACEi such as captopril and enalapril, and ARBs such as losartan, candesartan and irbesartan have been studied. These drugs would be expected to reduce portal pressure by reducing the Ang II mediated increase in intrahepatic resistance in cirrhosis. In addition to reducing Ang II production from Ang I, ACE inhibition also prevents the degradation of vasodilatory Ang-(1-7), and would be expected to increase intrahepatic Ang-(1-7) concentration. Although ACEi and ARBs are widely used in clinical practice to treat systemic hypertension, relatively limited and variable data are available to assess and compare their effectiveness in liver disease and portal hypertension[20,249-252].
A comprehensive meta-analysis by Tandon and colleagues[26]summarized the findings of nineteen clinical trials that included three ACEi and nine ARB studies performed up until 2009. This analysis concluded that ACEi and/or ARBs were equally effective as NSBBs in early Child Pugh A cirrhosis where ACEi/ARBs showed a similar reduction in HVPG (17%) to NSBBs (21%). However, in advanced Child Pugh B or C cirrhosis, ACEi/ARB’s were not effective in reducing HVPG and showed only a 3% reduction whereas NSBBs were just as effective as in Child Pugh A disease. This study concluded that in early stages of cirrhosis, the RAS is a major player in increased intrahepatic vascular resistance. However, in advanced cirrhosis, ACEi/ARBs are ineffective in reducing portal pressure likely due in part to the activation of other vasoconstrictive pathways such as sympathetic tone and the endothelin system that increase intrahepatic vascular tone[26,241,253]. Furthermore, it is known that treatment with ACEi/ARBs is associated with a significant hypotensive effect and renal impairment in patients with advanced cirrhosis because a baseline activation of the RAS is vital to maintain adequate arterial pressure and renal perfusion[26,254,255].
In addition, the effects of ACEi/ARBs as antifibrotic therapy have been studied in a number of clinical trials. In addition to any effects of these drugs on intrahepatic tone, attenuation of fibrosis would be expected to lead to improvement of intrahepatic vascular resistance to the portal blood flow. An open label randomized clinical trial reported that ARB candesartan significantly improved hepatic fibrosis in patients with compensated alcoholic cirrhosis with small but significant decrease in mean arterial pressure[256]. A recent systematic meta-analysis, which was performed on four randomized controlled trials published up until 2014, summarized the antifibrotic effects of ACEi/ARBs and suggested efficacy of these drugs in alleviating hepatic fibrosis but no data were available on the effects on portal pressure[257].
TARGETING THE ALTERNATE RAS IN PORTAL HYPERTENSION
As outlined above, there is considerable evidence that the components of the alternate RAS are upregulated in the hepatic, systemic and splanchnic circulations in cirrhosis[24,35,80,230]. It has been shown that vasodilatory actions of Ang-(1-7) is more prominent in the splanchnic vascular bed compared to the systemic vascular beds[258]. Indeed, in animal models, Ang-(1-7), the effector peptide of the alternate RAS, contributes to portal hypertension by reducing splanchnic vascular resistance, resulting in increased splanchnic blood flow[22]. The effects of Ang-(1-7) in splanchnic vascular bed appears to be mediated at least in partviathe MasR, because the specific MasR blocker A779 increased splanchnic vascular resistance, thereby reducing splanchnic blood flow in cirrhotic rat models[22]. However, this treatment produced only a modest effect on portal hypertension, particularly in one animal model where A779 also increased intrahepatic vascular resistance.
In addition to MasR, vasodilatory effects of Ang-(1-7) could also be mediated by the newly characterized MrgD[226]. Recent work by our laboratory demonstrated for the first time that similar to MasR, MrgD is also upregulated in the cirrhotic splanchnic vessels[23]. Moreover, a bolus injection of MrgD blocker D-Pro significantly reduced portal pressure in cirrhotic rat models (Figure 5A and B), and this reduction was similar to that of the MasR blockade. However, we noted that both MasR or MrgD blockers did not produce a clinically meaningful reduction in portal pressure (i.e., < 20% from the baseline) and the pressure lowering effect was only sustained for up to 20-25 min. This was possibly due to the rapid metabolism of these peptide blockers in the rat circulation (Figure 5C). Therefore, this study warranted further experiments in which continuous infusions of the receptor blockers should be employed to detect whether they have a clinically significant and meaningful effect on portal pressure in experimental models. Nevertheless, we made an important discovery in this study that unlike MasR, MrgD is not upregulated in the hepatic vascular bed of cirrhotic animals (Figure 6), suggesting that vasodilatory effects of MrgD may be confined to the splanchnic vascular bed in cirrhosis. This finding, if proven, is of central importance because MasR blockade with A779 did not lower portal pressure in the bile duct ligated cirrhotic model since the effect of A779 on hepatic MasR compromises the overall effect on portal pressure[22]. Thus, knowledge of the tissue-specific expression of MrgD is important since it could provide a guide for the development of novel therapies that specifically target splanchnic vasodilatation with minimal off-target effects.
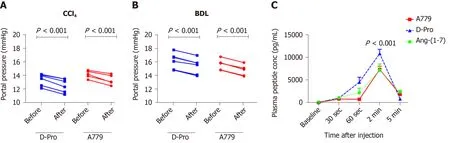
Figure 5 Portal pressure responses with Mas receptor and Mas-related G protein-coupled receptor type-D blockade and plasma angiotensin-(1-7) peptide (or blocker) concentrations.A and B: Portal pressure responses after systemic administration of a bolus dose of Mas receptor (MasR) blocker D-Ala7-Ang-(1-7) (A779) (10 mg/kg) or Mas-related G protein-coupled receptor type-D (MrgD) blocker D-Pro7-Ang-(1-7) (D-Pro) (10 mg/kg) in cirrhotic carbon tetrachloride-induced (A) and bile duct ligated (B) rats. Both MrgD and MasR blockade significantly reduced portal pressure likely by blocking angiotensin-(1-7) [Ang-(1-7)] mediated splanchnic vasodilatation; C: Plasma concentrations of Ang-(1-7) peptide and blockers measured before and after a bolus intra-jugular injection of Ang-(1-7) peptide (3.5 mg), MasR blocker A779 (3.5 mg) or MrgD blocker D-Pro (3.5 mg). The peptide and the blockers were injected into healthy rats and plasma levels of Ang-(1-7) peptide were measured by a radioimmunoassay using an antibody directed at middle amino acids of the peptide. Each time point represents the mean ± SEM profile from 4 rats per treatment group. P < 0.001, baseline vs 2-min post-injection and 2-min vs 5-min post-injection. Adapted from reference[23]. CCl4: Carbon tetrachloride-induced; BDL: Bile duct ligated; A779: D-Ala7-Ang-(1-7); D-Pro: D-Pro7-Ang-(1-7); Ang-(1-7): Angiotensin-(1-7).
CONCLUSION
In cirrhosis, portal hypertension is initiated by increased intrahepatic vascular resistance due to fixed obstruction of the portal vascular bed resulting from tissue fibrosis and other contractile cells. It is also associated with a hyperdynamic circulatory state characterized by a high cardiac output, increased total blood volume and splanchnic vasodilatation, resulting in increased mesenteric blood flow and this further increases portal pressure. Despite major advances in understanding the pathophysiology of portal hypertension, treatment options for this condition are limited. At present, NSBBs are the most widely accepted pharmacotherapy in clinical practice to prevent variceal bleeding. However, a significant proportion of cirrhotic patients are intolerant of NSBBs and fail to achieve an optimal therapeutic response. Therefore, there is an unmet major need to develop more safe, specific and effective pharmacotherapies for the treatment of portal hypertension.
Recent developments in understating the complex mechanisms of the pathogenesis of portal hypertension have opened up avenues for the development of novel therapies. These emerging therapeutic options target both the increased splanchnic vasodilatation and elevated intrahepatic vascular resistance. It is well known that the RAS is an important pathological system upregulated in both hepatic and splanchnic vascular beds in cirrhosis. Whilst the blockers of the classical RAS such as ACEi and ARBs can have a therapeutic effect by improving intrahepatic vascular toneviareduced hepatic fibrosis and vasoconstriction, they also cause adverse off-target effects such as systemic hypotension and renal failure. On the other hand, recent findings from studies that have investigated the potential use of the blockers of the components of the alternate RAS provided compelling evidence to justify the development of drugs targeting the splanchnic vascular bed to inhibit splanchnic vasodilatation in portal hypertension. In view of this, it is now evident that splanchnic vascular bed-specific inhibition of the alternate RAS may provide a better therapeutic option than manipulating the classical RAS in portal hypertension.
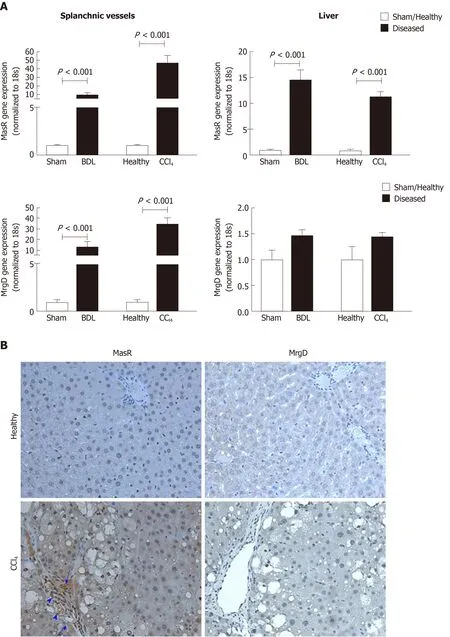
Figure 6 Expression of the receptors of the alternate renin angiotensin system in the splanchnic and hepatic vasculatures in cirrhosis.A: Gene expression of Mas receptor (MasR) and Mas-related G protein-coupled receptor type-D (MrgD) in cirrhotic mesenteric arterial vessels and livers of carbon tetrachloride-injected (CCl4) and bile duct ligated rats compared with sham-operated and healthy controls. Each bar represents the mean ± SEM profile from 6-7 rats per group. Gene expression of both MasR and MrgD are upregulated in the splanchnic vascular bed of both cirrhotic models, suggesting that both receptors are likely involved in angiotensin-(1-7)-mediated splanchnic vasodilatation in cirrhosis. In marked contrast, MasR, but not MrgD, is upregulated in the cirrhotic livers, suggesting that MasR, but not MrgD of the alternate renin angiotensin system, likely contributes to the regulation of hepatic vascular resistance in cirrhosis; B: Shows immunohistochemical localization of MasR and MrgD in the livers of CCl4 rats and healthy controls. Consistent with gene expression analysis, strong positive staining for MasR is shown in liver sinusoids (arrow), bile duct epithelial cells (arrowhead-large) and hepatic arterioles (arrowhead-small) of the cirrhotic liver. Consistent with gene expression analysis, there was no positive staining for MrgD in the cirrhotic liver. Adapted from reference[23]. MasR: Mas receptor; MrgD: Mas-related G proteincoupled receptor-type D; CCl4: Carbon tetrachloride-injected; BDL: Bile duct ligated.
 World Journal of Gastroenterology2020年40期
World Journal of Gastroenterology2020年40期
- World Journal of Gastroenterology的其它文章
- New strain of Pediococcus pentosaceus alleviates ethanol-induced liver injury by modulating the gut microbiota and short-chain fatty acid metabolism
- Prediction of clinically actionable genetic alterations from colorectal cancer histopathology images using deep learning
- Compromised therapeutic value of pediatric liver transplantation in ethylmalonic encephalopathy: A case report
- Predicting cholecystocholedochal fistulas in patients with Mirizzi syndrome undergoing endoscopic retrograde cholangiopancreatography
- Novel endoscopic papillectomy for reducing postoperative adverse events (with videos)
- Pediatric bowel preparation: Sodium picosulfate, magnesium oxide, citric acid vs polyethylene glycol, a randomized trial