
进化树在细菌亲缘关系分析中的应用研究
2021-01-04 03:28迟文静刘宜昕张艳梅
检验医学 2020年12期
迟文静, 刘宜昕, 王 粟, 刘 涛, 赵 虎, 张艳梅
(复旦大学附属华东医院检验科,上海 200040)
分子系统发育学是利用分子特征研究生物体之间进化关系的学科,在揭示生物进化途径、研究生物多样性和分子流行病学特征、鉴定菌种和基因功能等方面发挥重要作用[1]。早期微生物分子系统发育研究依赖于蛋白质序列,其中应用广泛的蛋白质序列有铁氧还蛋白和细胞色素等。20世纪70年代中期,有学者开始使用16SrRNA的基因序列构建进化树,以确定不同生物之间的进化关系。目前,随着高通量测序(next generation sequencing,NGS)等技术的应用,大大降低了基因分析的成本,加快了基因分析的速度,为细菌进化树的构建提供了更多类型的分析和展示形式[2]。本文对进化树及其在细菌亲缘关系中的应用进行综述。
1 进化树概述
1.1 进化树定义
进化树又称系统发生树,是描述生物体形成或进化顺序的拓扑树结构,通常是二叉树的形状,一般由一系列节点和分支组成,节点代表某个具体序列,节点之间的连线代表物种之间的亲缘关系[3]。构建进化树不仅需要分子生物学、遗传学、生态学等生命科学学科知识,还需要统计学、计算机学等多个学科知识的融合[4]。微生物进化树的构建过程主要有:序列数据的获取、进化距离模型的确定、多个序列的比对、对比后结果的提取以及算法和参数的选择[5]。进化树可以分析未知细菌和已知细菌间的亲缘关系[6],在遗传本质上探究细菌多样性的产生机制。
1.2 进化树的呈现方式
根据是否制定根节点,进化树可以分为有根树和无根树2种呈现方式[7]。有根树制定了根节点,从树中可以看出各节点之间的距离和各分支分化的先后关系。有根树引入外群作为根节点,而外群通常选择与研究序列关系密切的序列,且能很好地聚类;或者选择比研究序列进化历史更早的序列,故有根树可以看到不同细菌间关系的远近,还可以看到细菌的进化顺序和方向[8]。无根树不引入外群,没有根节点,只能看出个各个节点的拓扑结构和相对距离,因而无根树功能单一,可以显示不同细菌的聚类关系和相对距离的远近,却无法显示细菌的起源和进化方向[9]。见图1。
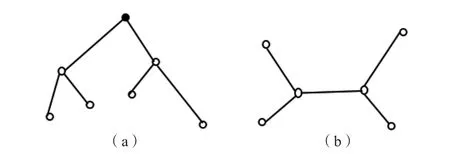
图1 进化树的呈现方式
1.3 进化模型
进化模型是对微生物进化变异进行的数学描述,描述内容主要有:基因的点突变、插入缺失,各碱基突变发生的概率及核苷酸组成频率等和目标菌株间的亲缘关系,选择和构建合适的进化模型是研究细菌进化的前提[10]。生物进化的研究按层次可分为宏进化和微进化。宏进化是细菌间的进化,主要指细菌不同目/科/属/种的进化[11],微进化是指细菌种内或近缘菌种间的进化[12]。以幽门螺杆菌为例,在其微进化过程中,构建幽门螺杆菌不同菌株间的进化树模型,不仅需要估计碱基点突变率,还需要估计重组率、核苷酸差异率及重组核苷酸片段长度等重要信息[13]。对于细菌的进化分析,选择合适的进化模型才可能得到可靠的结果,反之可能得到不准确甚至是错误的结果,这种现象主要是由进化树不相同的拓扑结构或分支长度造成的,因此选择合适的进化模型、构建正确的进化树至关重要。
1.4 进化树分析方法
1.4.1 距离矩阵法 距离矩阵法是一种以细菌核苷酸序列间的变异估计菌株间距离,并通过距离矩阵构建进化树的方法。这种方法首先需要将输入的核苷酸序列数据转化为距离信息,然后通过距离信息进一步构建进化树,主要分为系统树法和网络法[14]。总的来说,距离矩阵法是一种基于距离构建进化树的方法,其优点在于简单、直观、计算速度快,但在菌株间进化速率差异较大的情况下,可能得到错误的拓扑结构[15]。
1.4.2 最大简约法 最大简约法是一种将细菌的核苷酸序列位点视为形状的方法,是一个比较所有可能的拓扑结构的过程。最大简约法首先筛选出对细菌进化分析有用的核苷酸位点,然后统计每个位点的核苷酸最小替换数,进而以各位点替代数总和最小的进化树作为最优树[16]。相对于距离矩阵法,最大简约法对信息的利用度更高,而相对于极大似然法和后验概率法,该方法计算速度更快,可处理较多的插入、缺失序列。
1.4.3 极大似然法 极大似然法是利用进化模型和核苷酸序列,通过进化树的分枝长度、拓扑结构、模型参数构建进化树的方法。极大似然法通过选取合适的进化模型分析核苷酸序列,得到似然率最大的拓扑结构,再以其中最大似然率的拓扑结构构建的进化树作为最优树,应用极大似然法分析比较重要的问题时需要确定最合适的进化模型[17]。一般在选择合理、正确的进化模型的情况下,极大似然法可以推导出很好的进化树结果,但与最大简约法相比,极大似然法很难在序列长度较短的情况下得到正确的拓扑结构。
1.4.4 后验概率法 后验概率法又被称为贝叶斯推论法,该方法首先假定所有可能的进化树是等概率的,然后计算出比对后序列的进化树的后验概率,进而将后验概率最大的进化树作为最优树[18]。后验概率法的优点在于不但具有数学和统计学基础,同时还可以处理复杂的、接近实际情况的进化模型。与极大似然法相比,后验概率法同样应用广泛,且可通过相同的数据信息,更低的计算量,得出与极大似然法一致的结论。与最大简约法相比,后验概率法能够考虑更多的进化相关信息。
2 进化树的构建
2.1 基于保守基因构建进化树
2.1.1 基于核心基因组多位点序列分型(multilocussequence typing,MLST)构建进化树 核心基因序列是重复且保守的核苷酸序列,在进化过程中可能发生富集、选择和遗传[19]。细菌的核心基因序列之间可以通过直接接触及质粒、噬菌体或其他可移动遗传元件(整合子、转座子和插入序列等)的方式进行基因复制或基因交换,通过分析这些保守序列,可以分析不同细菌进化的差异以及菌株间的亲缘关系[20]。MLST是近年来发展迅速的分子生物学分析方法,具有较高的分辨能力,可以通过多个管家基因450 bp左右的基因序列比较菌株等位基因的多态性,不同菌株对应不同的序列型,可以进行菌株进化和种群结构的研究[21]。核心基因组MLST可以使用微生物数百甚至数千个保守的等位基因进行基因分型,其分辨率远高于传统的MLST[22]。随着NGS技术的广泛应用,核心基因组MLST主要被应用在分子流行病学分析领域,且在该领域快速发展,已有研究采用核心基因组MLST方法分析我国即食食品中单核细胞增生李斯特菌的分子流行病学特征,结果表明核心基因组MLST能将不同谱系、血清群和克隆群的菌株明显分开,共分为24个亚群,与克隆群基本保持一致[23]。基于全基因组测序的核心基因组MLST分辨能力强,可用于监测暴发性食源性疾病。但核心基因组MLST仍有一定的局限性,如缺乏对关系非常密切的菌株的分辨能力,且可分析菌株的种类有限[24]。2011年,SAHL等[25]对大肠埃希菌的MLST研究结果显示,全基因组序列进化树与MLST分型的结果并不一致。2014年,朱健铭等[26]对肺炎克雷伯菌进行分析,结果表明采用单个核苷酸序列进行细菌进化分析及采用MLST进行菌株亲缘关系分析并不可靠,为了校正MLST的缺点,提高分辨率,他们采用管家基因和毒力基因联合检测的方法进行分析,发现根据核心基因组各位点序列图谱构建的进化树,与参与菌株相应序列进行比对,既可以准确地研究细菌遗传进化关系,又可以确定菌株的种属,对分析细菌亲缘关系意义重大。
2.1.2 基于非编码保守基因构建进化树 非编码保守DNA序列(conserved noncoding DNA sequences,CNS)是指细菌基因组中转录RNA但不能翻译蛋白质或能调控其他基因的序列,是比较小的一段序列[27]。CNS在生物的进化中具有加工修饰RNA、调控转录和DNA结构等特殊的功能[28]。CNS不仅具有长度和频率的物种特异性,还具有丰富性、广阔性、保守性和功能性等特点,使其在微生物亲缘关系的分析中极具潜力,如对耐辐射球菌与嗜热菌亲缘关系的分析[29]。但是如果要对CNS的功能进行正确、全面的理解和分析,还需要通过大量的实验进行功能验证。
2.2 基于单核苷酸多态性(single nucleotidepolymorphism,SNP)构建进化树
SNP是指基因组中的单个碱基的突变引起的DNA序列多态性,有数量多、多态性丰富、遗传稳定、易实现分析自动化的特点[30]。单个碱基的变异可以由颠换或转换引起,也可以由插入或缺失引起,这些SNP位点可能影响基因的功能,引起性状的改变,甚至导致疾病的发生,因此SNP是遗传变异的重要依据,被广泛应用于微生物的起源、进化及迁移等方面的研究。有学者为了解外源基因转化沙漠寡营养细菌的进化与变异,通过生物学信息绘制了细菌的SNP系统发育树,结果表明SNP数量最多的菌株进化速度最快[31]。SNP分析不仅提供了研究和理解基因突变的新方法,还能为微生物的鉴定及亲缘关系分析提供依据。
2.3 基于基因拷贝数构建进化树
基因拷贝数是某种基因或某段特定DNA序列在基因组中出现的数目,多拷贝基因广泛存在于细菌中,而且多是可以移动的基因序列,分析基因拷贝数变异(jcopy number variation,CNV)是研究微生物进化、变异以及致病性的基础[32]。CNV是基因组中的多核苷酸突变,是一种重要的遗传变异,与研究较多的SNP相比,CNV涉及更多的碱基,覆盖更大范围的基因序列,在基因突变与细菌进化的研究上逐渐成为热点和重点[30]。目前,全基因组CNV检测的方法主要有芯片法和NGS技术等。全基因组CNV图谱和更精确的参考基因组必将引领微生物基因组学研究热潮,并进一步提高在全基因组范围内探测基因组变异的准确性,对微生物亲缘关系的分析有重大的指导意义。
3 基于其他特征基因构建进化树
3.1 基于致病岛构建进化树
致病岛又被称为毒力岛、适应岛、生态岛或共生岛,是细菌基因组中可以编码毒力因子的序列,与细菌的致病性密切相关,能够在菌株间通过基因组水平转移,可以使细菌在短期内发生形状的改变,甚至产生新的变种,这种演变有助于细菌不断适应环境[33]。很多病原菌都有致病岛,如产肠毒素葡萄球菌、幽门螺杆菌、大肠埃希菌、沙门菌等[34-35]。基于致病岛构建进化树有助于理解细菌的进化和迁徙。有学者[36]研究了基于幽门螺杆菌CagPAI和Cag A基因构建的系统发育树,验证了拉丁美洲菌株的分群和聚类特点,以及与之相关的人群背景。随着微生物耐药性的增强和新型病原体的出现,鉴定致病菌也越来越重要。鉴定病原体的毒力基因并了解其从非致病性向致病性的进化,对于基础科学和医学研究都是一种挑战。
3.2 基于规律成簇间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)构建进化树
CRISPR在细菌基因组中由不连续的同向重复序列和插入其中的间隔序列组成,有针对噬菌体或质粒等外源基因的获得性免疫作用,在细菌进化过程中保持结构的高度可变,CRISPR位点是研究细菌分型与进化的关键位点[37]。CRISPR通常由同向重复序列、间隔序列、前导序列以及CRISPR相关蛋白组成。cas基因与重复序列相互关联,可使细菌协同进化[38]。CRISPR位点会随着细菌的进化不断出现新间隔序列的插入以及旧间隔序列的丢失,这种现象是导致细菌基因组进化速度快的重要原因之一,因此CRISPR位点具有很复杂的多态性[39]。CRISPR位点在细菌中的多态性不仅能够反映细菌与环境相互作用的关系,还能够记录细菌在进化过程中的生态学和地理学信息,如根据CRISPR位点的间隔序列在沙门菌中排列的差异,可以判断不同菌株间的亲缘关系,并进行溯源分析;根据醋酸菌的重复序列构建进化树,可将不同属的菌种进行分类[40]。有研究应用CRISPR序列分型分析病原体的暴发流行,如鼠疫耶尔森菌和肠炎沙门菌亚种的分群[40-41]。此外,CRISPR还可以提供与微生物表型相关的重要信息,如肠球菌耐药基因序列和化脓性链球菌基因组中的前噬菌体等,这些信息都反映了CRISPR在调控基因水平转移、细菌适应环境及细菌进化中的作用[42]。因此,应用CRISPR构建进化树分析细菌亲缘关系的优点在于:基因分型分辨率较高、操作简单、重复性好、结果数字化、便于不同实验室结果的比对等[43]。但目前仍然存在一些需要解决的问题,如数据不够充分、数据库不够健全、细菌之间的分型标准不够完善等,在未来,构建标准化的CRISPR数据库具有十分重要的意义。
4 总结和展望
基因分析技术的飞速发展促进了生物学诸多领域的发展,对认识细菌基因组与进化的关系有深远的影响。细菌基因组具有多样性和规律性特征,面对海量的基因组信息,挖掘有效信息,构建合适的进化树是十分重要的。有效地构建进化树应该具备2个条件:首先,必须基于一种合适的进化方式,能反映或解释进化事件;其次,应该覆盖更多的基因组信息[44]。为更好地鉴定细菌类型以及明确不同菌群之间的亲缘关系,结合不同基因组序列构建细菌进化树,将有效帮助解决细菌进化中许多悬而未决的问题[45]。
本文简述了目前常见的用于构建细菌进化树的方法,但是可以用来进行细菌亲缘关系分析而构建进化树的方法不限于文中所述,挖掘更多有价值的基因标志物将是很有前景的研究,也是探究细菌进化史的必由之路。
猜你喜欢
作物学报(2022年2期)2022-11-06
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
天津市教科院学报(2021年5期)2021-11-10
生物学通报(2021年9期)2021-07-01
中国饲料(2019年19期)2019-03-25
天然产物研究与开发(2018年10期)2018-11-06
广东农业科学(2017年5期)2017-08-29
中国医学创新(2017年7期)2017-03-31
