
基于2′,4′-二氟联苯基的新型1,2,4-三氮唑类化合物的合成及其抗真菌活性
2020-12-29 08:46魏文博段坤坤方家宝张尹超杨占涛
华中师范大学学报(自然科学版) 2020年6期
陈 勇,魏文博,段坤坤,方家宝,张尹超,杨占涛
(安阳师范学院化学化工学院, 河南 安阳 455000)
真菌感染是十分常见的疾病,它能够造成皮肤、粘膜等浅表皮处的感染,这类真菌感染中最常见的是皮肤癣菌病.真菌也可以造成皮下组织等深部感染,可侵犯内脏及脑膜等处,甚至引起全身播散性感染,既侵袭性真菌感染.引起的疾病如真菌性肠炎,脑膜炎等,严重的状况下,会造成人的死亡.近年来,随着高效广谱抗生素、免疫抑制剂、抗恶性肿瘤药物的广泛应用,器官移植、体内置管以及久驻ICU病人的增加,侵袭性真菌感染引起的发病率和致死率逐年上升,已成为危害人类健康的严重威胁之一[1-6].当前,2019新型冠状病毒(Severe Acute Respiratory Syndrome Coronavirus 2,简称SARS-CoV-2)引起的新型冠状病毒肺炎(Corona Virus Disease 2019,简称COVID-19) 仍在全球肆虐并引起了全球性关注,新型冠状病毒肺炎传染性强,发病率高,重症患者病死率高.重症新冠肺炎患者具备侵袭性真菌感染发生的高危因素,且已有重症病例出现继发或合并真菌感染报告[7].药物治疗是应对侵袭性真菌感染的主要方法,然而已有的抗真菌药物种类有限,致病性真菌对这些药物产生耐药性,大部分药物还具有毒副作用等问题,使得研发新的抗真菌药物成为必要.
氮唑类抗真菌药物是目前常用的一类抗真菌药物,按照药物的分子结构可分为咪唑类和三氮唑类.氮唑类药物的作用机制:通过抑制ERGl1基因编码的14α-羊毛甾醇脱甲基酶(CYP51),抑制真菌细胞膜中的羊毛甾醇转化为麦角甾醇,并使毒性的甾醇产物在真菌细胞中积累,从而抑制真菌的生长与复制[8-9].咪唑类抗真菌药物(咪康唑,酮康唑等)是一类开发较早,抗真菌活性较高,但毒副作用较大,主要用于浅表真菌感染的治疗.后来科研工作者研制出了三氮唑类抗真菌药,相对于咪唑类抗真菌药,三氮唑类抗真菌药具有毒性较低,抗菌谱较广,可用于治疗深部真菌感染引起的疾病.但是三氮唑类抗真菌药久用易使真菌产生耐药性,药效渐弱.为此研究者通过不断的改进药物分子的结构,来应对致病性真菌的耐药性,进而提高三氮唑类抗真菌药的疗效.研究者先后研制出了第一代三氮唑类抗真菌药氟康唑、伊曲康唑等;第二代三氮唑类抗真菌药伏立康唑、沙伯康唑等;最新上市的三氮唑类抗真菌药艾沙康唑[9-11].
目前临床上可用于治疗深部真菌感染的药物除三唑类抗真菌药物外,还有多烯类抗真菌抗生素两性霉素B(Amphotericin B)、制霉菌素A1(Nystatin A1);棘白菌素类抗真菌药卡泊芬净(Caspofungin)、米卡芬净(Micafungin)[12-14].三唑类抗真菌药物作为一类全合成化学药,可利用有机合成的多样性,用有机合成的方法,合成种类繁多,结构新颖的具有潜在抗真菌活性的化合物,进而可在此基础上开发出高效、低毒、广谱的抗真菌药物.因此,三唑类抗真菌药仍是目前临床上用于治疗深部真菌感染的主要药物.
尽管科研工作者已经开发了多个分子结构不同的三唑类抗真菌药物,但由于多种原因导致的真菌感染的疾病也在增多,而现有的抗真菌药物也存在一定的毒副作用,尤其是真菌耐药性的增加,所有这些均给真菌感染疾病的治疗带来困难.因此开发新型高效、低毒的抗真菌药物仍有必要[15-18].
为了应对致病性真菌对现有药物产生的耐药性,并提高药物的药效性,一个有效的方法是对原有药物的分子结构进行化学修饰[19-21].已用于临床的三唑类抗真菌药多以2,4-二氟苯基为分子骨架,通过不断地化学修饰,分别开发出了一、二、三代三唑类抗真菌药[8-9].从药物开发的实践看,联苯基是构成药物分子的理想结构片段,根据药物分子设计原理中的结构骨架变换原则[22],新药的创制是在发现活性分子基础上的骨架变换.因此,本研究以已有的唑类抗真菌药物的构效关系为基础,依据药物分子设计原理,设计并合成以 2,4-二氟代联苯基为分子骨架的新型三氮唑化合物.合成路线如Scheme 1所示.
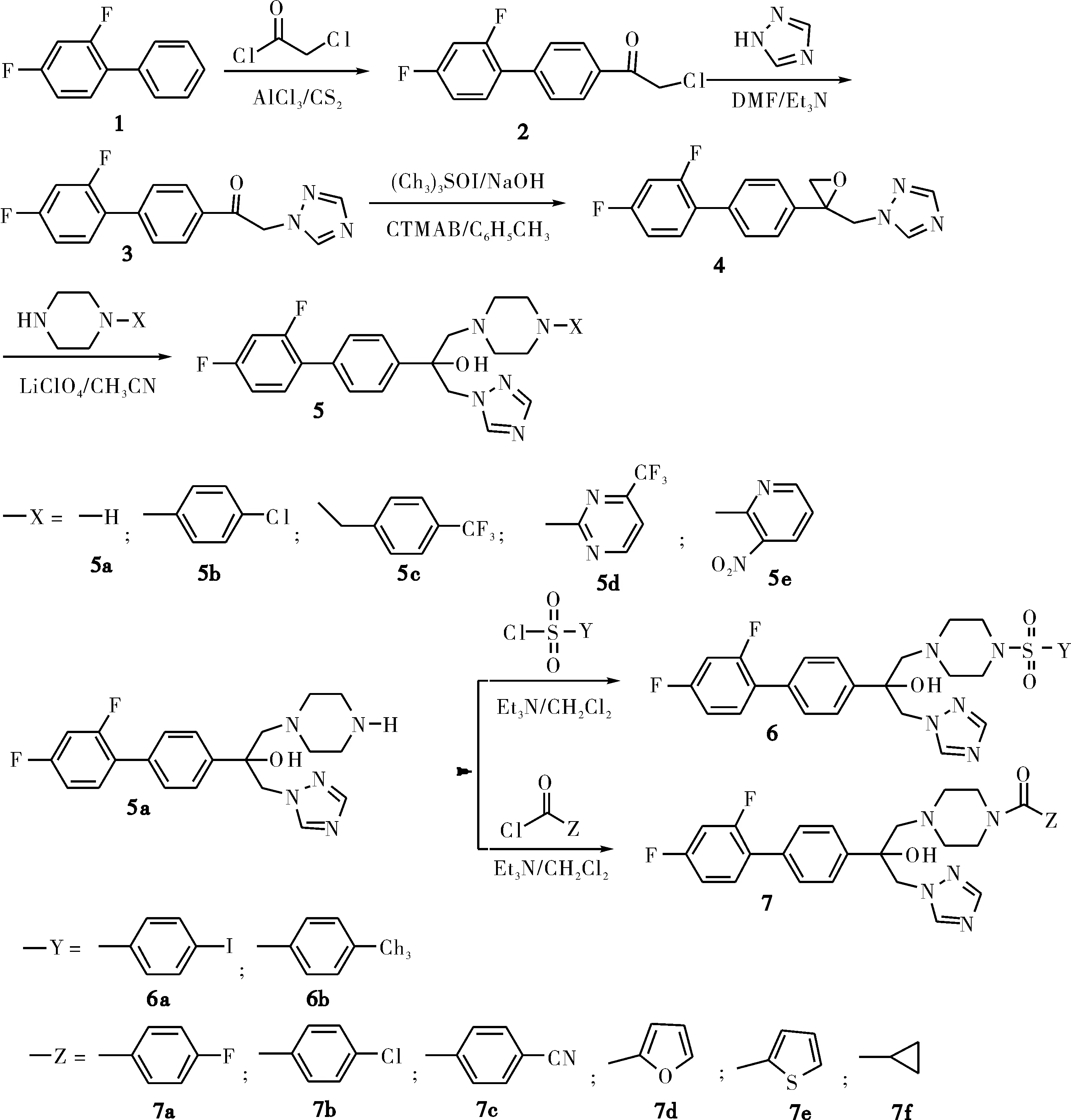
Scheme 1 目标化合物的合成路线 Scheme 1 The synthetic route of the title compounds
1 实验部分
1.1 仪器和试剂
ACQUITYTMUPLC/LCT Premier XE型高分辨液相质谱仪(美国Waters公司);Am-400型核磁共振仪(德国Bruker公司).
试剂:2,4-二氟联苯、 三甲基碘化亚砜、十六烷基三甲基溴化铵、CDCl3、d6-DMSO(分析纯,阿拉丁)、1-(4-三氟甲基苯甲基)哌嗪、高氯酸锂、1-(4-三氟甲基嘧啶-2-基)-哌嗪,1-(3-硝基吡啶-2-基)-哌嗪、4-碘苯磺酰氯、4-甲基苯磺酰氯、对氟苯甲酰氯、对苯甲酰氯、对氰基苯甲酰氯、2-呋喃甲酰氯、2-噻吩甲酰氯、环丙基(哌嗪-1-基)甲酮、三乙胺(分析纯,阿拉丁);四氢呋喃、乙酸乙酯、石油醚、乙腈、二氯甲烷(分析纯,科密欧);柱层析硅胶(青岛海洋化工厂)54~57 μm.
1.2 实验方法
1.2.1 目标化合物的合成
1) 1-[2′,4′-二氟-(1,1′-联苯基)-4-基]-2-氯乙酮 (2) 和1-(2′,4′-二氟联苯-4基)-2-(1H-1,2,4-三氮唑-1-基)-乙酮(3)的合成同文献[23].
2) 1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-2,3-环氧丙烷(4)的合成向反应瓶中加入1.55 g (5.19 mmol) 化合物3、1.26 g (5.72 mmol) 三甲基碘化亚砜、0.19 g十六烷基三甲基溴化铵、13 mL的甲苯和2 mL NaOH(aq. 20 %),控温60 ℃,搅拌反应1.5 h.向反应后的混合溶液加入25 mL乙酸乙酯,转入分液漏斗,混合液用水洗涤,洗涤后的有机层用无水硫酸镁干燥.减压蒸去溶剂,余物用石油醚:乙酸乙酯(V∶V=1∶1)的展开剂过硅胶色谱柱,得到0.89 g棕红色粘稠状化合物4,产率54.9 %.
HMRS-ESI:m/z[M+H] calcd. for C17H14F2N3O: 314.1105; found: 314.1112.
3) {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯基-4-基)-3-(-1-哌嗪基)}-2-丙醇(5a)的合成
把11.25 mL的乙醇和11.25 mL的THF加入反应瓶中,摇匀,配成混合溶剂;再向反应瓶中加入0.48 g (5.6 mmol)哌嗪、1.17 g (3.75 mmol)化合物4和1.25 ml的三乙胺,在65 ℃下搅拌反应 7 h.反应结束后,向反应瓶中加入25 mL蒸馏水,把反应瓶中的混合液转入分液漏斗,混合液用25 mL×2乙酸乙酯萃取,萃取液用无水硫酸镁干燥,旋蒸除去溶剂,余物过硅胶色谱柱,先用乙酸乙酯作洗脱剂,把少量杂质先行洗脱除去,待产物流出后,改用甲醇作洗脱剂,得1.11 g白色固体产物化合物5a,产率 88.5%.
HMRS-ESI:m/z[M+H] calcd. for C21H24F2N5O: 400.1949; found: 400.1942.
4) {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-氯苯基)-1-哌嗪基]}-2-丙醇(5b)的合成
称取0.106 7 g (0.34 mmol )化合物4,0.098 3 g(0.5 mmol )1-[(4-氯苯基)]-哌嗪,0.106 g (1 mmol)LiClO4及4 mL乙腈加入 到反应瓶中,在85 ℃下,搅拌18 h.然后,向反应混合物中加入10 mL蒸馏水并用乙酸乙酯(15 mL×2)萃取,合并有机相,无水硫酸镁干燥,旋蒸除去有机溶剂,余物进行硅胶色谱分离,洗脱剂为石油醚:乙酸乙酯(V∶V)=1∶1,得0.116 g橙红色蜡状物化合物5b,产率为67.1%.
1H NMR (400 MHz,CDCl3) δ 8.14 (s,1H),7.90 (s,1H),7.49 (d,J=29.6 Hz,4H),7.42 (dd,J=15.2,8.6 Hz,1H),7.17 (d,J=8.9 Hz,2H),6.95 (dt,J=19.2,8.6 Hz,2H),6.75 (d,J=8.9 Hz,2H),4.84 (s,1H),4.48 (d,J=14.3 Hz,1H),4.37 (d,J=14.3 Hz,1H),3.06 (s,1H),3.01 (t,J=4.8 Hz,4H),2.77 (d,J=13.4 Hz,1H),2.52 (dd,J=10.4,5.3 Hz,2H),2.44 (dd,J=10.5,5.2 Hz,2H).
13C NMR (101 MHz,CDCl3) δ 163.64,161.09,158.54,151.17,149.56,144.88,143.07,134.33,131.33,129.08,128.94 (2C),125.35 (2C),124.77,117.20 (2C),111.75,111.55,104.45,73.02,63.66,58.07,54.21 (2C),49.21 (2C).
HMRS-ESI:m/z[M+H] calcd. for C27H27ClF2N5O: 510.1872; found: 510.1883.
5) 化合物5c~5e的合成方法和化合物5b的合成方法相同.
① {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-三氟甲基苯甲基)-1-哌嗪基]}-2-丙醇(5c)的合成
橙色蜡状产物,产率 45.0%.
1H NMR (400 MHz,CDCl3) δ 8.13 (s,1H),7.88 (s,1H),7.61~7.46 (m,6H),7.43~7.35 (m,3H),7.00~6.86 (m,2H),4.43 (d,J=14.3 Hz,1H),4.33 (d,J=14.3 Hz,1H),3.85 (s,1H),3.52 (d,J=13.5 Hz,1H),3.44 (d,J=13.5 Hz,1H),2.99 (d,J=13.3 Hz,1H),2.72 (d,J=13.3 Hz,1H),2.38 (s,2H),2.32 (s,6H).
13C NMR (101 MHz,CDCl3) δ 163.65,161.15,158.35,151.21,149.66,144. 81,143.05,134.23,131.38,129.12,128.97 (2C),125.29 (2C),124.81,123.78,117.23 (2C),111.63,111.56,104.48,73.24,65.39,64.59,58.12,54.37 (2C),48.22 (2C).
HMRS-ESI:m/z[M+H] calcd. for C29H29F5N5O: 558.2292; found: 558.2285.
② {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-三氟甲基嘧啶-2-基)-1-哌嗪基]}-2-丙醇(5d)的合成
黄色蜡状物,产率56.4%.
1H NMR (400 MHz,CDCl3) δ 8.43 (d,J=4.7 Hz,1H),8.14 (s,1H),7.89 (s,1H),7.52 (s,4H),7.41 (dd,J=15.2,8.5 Hz,1H),6.94 (dt,J=19.2,8.6 Hz,2H),6.72 (d,J=4.8 Hz,1H),4.91 (s,1H),4.48 (d,J=14.3 Hz,1H),4.38 (d,J=14.3 Hz,1H),3.73 (s,4H),3.03 (d,J=13.4 Hz,1H),2.77 (d,J=13.4 Hz,1H),2.42 (dd,J=10.7,5.4 Hz,2H),2.34 (dd,J=10.6,5.3 Hz,2H).
13C NMR (101 MHz,CDCl3) δ 163.63,161.13,160.02,158.53,156.20,151.17,144.89,142.98,134.38,131.34,129.09,128.89,125.32(2C),121.87,119.13,111.73,111.52,104.87,73.13,63.90,58.01,54.13 (2C),43.65 (2C).
HMRS-ESI:m/z[M+H] calcd. for C26H25F5N7O: 546.2041; found: 546.2035.
③ {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(3-硝基吡啶-2-基)-1-哌嗪基]}-2-丙醇(5e)的合成
橙红色蜡状物,产率为61.0%.
1H NMR (400 MHz,CDCl3) δ 8.31~8.24 (m,1H),8.13 (s,1H),8.08 (dd,J=8.0,1.5 Hz,1H),7.89 (s,1H),7.51 (s,4H),7.47~7.36 (m,1H),7.03~6.88 (m,2H),6.73 (dd,J=8.0,4.5 Hz,1H),4.84 (s,1H),4.47 (d,J=14.3 Hz,1H),4.37 (d,J=14.3 Hz,1H),3.45~3.32 (m,2H),3.33~3.22 (m,2H),3.05 (d,J=13.4 Hz,1H),2.76 (d,J=13.4 Hz,1H),2.52~2.43 (m,2H),2.43~2.32 (m,2H).
13C NMR (101 MHz,CDCl3) δ 163.63,161.09,158.53,152.54,151.69,151.19,144.89,142.90,135.51,134.40,133.16,131.36,129.12,129.10,125.30 (2C),113.68,111.62,104.43,73.13,63.80,58.00,54.04 (2C),47.93 (2C).
HMRS-ESI:m/z[M+H] calcd. for C26H26F2N7O3: 522.2065; found: 522.2071.
6) {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-碘苯磺酰基)-1-哌嗪基]}-2-丙醇(6a)的合成
把399 mg(1 mmol)化合物5a、303 mg(1 mmol)4-碘苯磺酰氯、15 mL新蒸的二氯甲烷和320 μL三乙胺加入到反应瓶中.室温条件下磁力搅拌9 h.反应结束后,将反应混合物旋蒸除去溶剂,余物过硅胶色谱柱,洗脱剂为乙酸乙酯,浓缩后得到245 mg黄色固体化合物6a,产率为36.8%.
1H NMR (400 MHz,CDCl3) δ 8.05 (s,1H),7.90 (d,J=8.4 Hz,2H),7.86 (s,1H),7.51~7.35 (m,7H),7.01~6.87 (m,2H),4.50 (s,1H),4.41 (d,J=14.4 Hz,1H),4.30 (d,J=14.4 Hz,1H),2.99 (d,J=13.5 Hz,1H),2.88 (s,4H),2.69 (d,J=13.6 Hz,1H),2.50~2.42 (m,2H),2.42~2.30 (m,2H).
13C NMR (101 MHz,CDCl3) δ 163.65,161.07,158.52,151.25,149.56,144.85,142.52,138.43(2C),134.47,131.3(2C),129.04(2C),125.23 (2C),124.36,111.78,104.48,100.60,74.58,63.42,57.74,53.44 (2C),45.90 (2C).
HMRS-ESI:m/z[M+H] calcd. for C27H27IF2N5O3S: 666.0847; found: 666.0853.
7) {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-甲基苯磺酰基)-1-哌嗪基]}-2-丙醇(6b)的合成方法和化合物(6a)相同.
黄色蜡状固体0.209 6 g,产率47.4%.
1H NMR (400 MHz,CDCl3) δ 8.05 (s,1H),7.87 (s,1H),7.59 (d,J=8.2 Hz,2H),7.44 (dt,J=14.1,7.4 Hz,5H),7.34 (d,J=8.0 Hz,2H),6.95 (ddd,J=19.3,9.3,7.2 Hz,2H),4.56 (s,1H),4.40 (d,J=14.4 Hz,1H),4.29 (d,J=14.4 Hz,1H),3.00 (d,J=13.5 Hz,1H),2.87 (s,4H),2.69 (d,J=13.5 Hz,1H),2.46 (s,3H,—CH3),2.45~2.32 (m,4H).
13C NMR (101 MHz,CDCl3) δ 163.64,161.11,158.48,151.28,149.51,144.75,142.62,139.54,134.47,132.38(2C),131.34(2C),129.04(2C),125.23 (2C),124.36,111.78,104.48,74.58,63.42,57.74,53.44 (2C),45.90 (2C),24.58 (—CH3).
HMRS-ESI:m/z[M+H] calcd. for C28H30F2N5O3S: 554.2037; found: 554.2044.
8) {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-氟苯甲酰基)-1-哌嗪基]}-2-丙醇(7a)的合成
称取100 mg (0.25 mmol) 化合物5a、39.6 mg(0.25 mmol) 对氟苯甲酰氯、3 mL新蒸的二氯甲烷和50.6 mg (0.5 mmol) 三乙胺加入到反应瓶中,在磁力搅拌下,室温反应8 h.待反应结束后,反应混合液旋蒸除去溶剂,余物过硅胶色谱柱,洗脱剂为纯乙酸乙酯,得到72 mg灰白色固体化合物7a,产率为55.3%.
1H NMR (400 MHz,CDCl3) δ 8.17 (s,1H),8.08 (s,1H),7.91 (s,1H),7.43~7.28 (m,4H),7.11 (t,J=8.6 Hz,1H),7.05 (t,J=8.5 Hz,2H),6.92 (m,3H),5.51 (s,1H),4.48 (d,J=14.4 Hz,1H) 4.37 (d,J=14.4 Hz,1H),3.76~3.27 (m,4H),3.04 (d,J=13.4 Hz,1H),2.76 (d,J=13.4 Hz,1H),2.49~2.28 (m,4H).
13C NMR (101 MHz,CDCl3) δ 169.52,164.78,163.74,161.01,158.64,151.11,142.71,134.58,132.62,132.53,131.42(2C),129.13(2C),125.35 (2C),124.45,115.63(2C),111.87,104.56,77.36,73.31,63.82,58.03 (2C),53.99 (2C).
HMRS-ESI:m/z[M+H] calcd. for C28H27F3N5O2: 522.2117; found: 522.2109.
9) 化合物7b~7f的合成方法和化合物7a的合成方法相同.
① {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-氯苯甲酰基)-1-哌嗪基]}-2-丙醇(7b)的合成
棕色蜡状固体,产率为41.6%.
1H NMR (400 MHz,CDCl3) δ 8.13 (s,1H),7.89 (s,1H),7.48 (s,4H),7.42~7.32 (m,3H),7.28 (d,J=2.1 Hz,2H),6.97~6.86 (m,2H),4.47 (d,J=14.4 Hz,1H),4.36 (d,J=14.4 Hz,1H),3.73 (s,1H),3.40 (d,J=105.0 Hz,4H),3.02 (d,J=13.4 Hz,1H),2.75 (d,J=13.5 Hz,1H),2.47 ~2.17 (m,4H).
13C NMR (101 MHz,CDCl3) δ 169.20 (C=O),163.62,161.05,158.48,151.12,144.89,142.64,135.93,134.46,133.67,131.27,129.13(2C),128.77(2C),128.54(2C),125.25 (2C),124.35,111.67,104.46,73.25,63.74,57.87 (2C),54.31 (2C).
HMRS-ESI:m/z[M+H] calcd. for C28H27ClF2N5O2: 538.1821; found: 538.1827.
②{1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(4-氰基苯甲酰基)-1-哌嗪基]}-2-丙醇(7c) 的合成
棕色蜡状物固体产物,产率50.8%.
1H NMR (400 MHz,CDCl3) δ 8.13 (s,1H),7.89 (s,1H),7.72 (d,J=8.3 Hz,1H),7.65 (d,J=8.2 Hz,1H),7.47 (s,3H),7.42 (d,J=8.2 Hz,1H),7.39~7.32 (m,1H),6.99~6.84 (m,1H),5.12 (s,1H),4.42 (dd,J=47.0,14.4 Hz,2H),3.65 (d,J=71.0 Hz,2H),3.21 (s,1H),3.02 (d,J=13.5 Hz,1H),2.75 (d,J=13.5 Hz,1H),2.35 (dd,J=81.2,31.8 Hz,3H).
13C NMR (101 MHz,CDCl3) δ 168.16 (C=O),163.61,161.04,158.45,150.96,144.90,142.50,139.69,134.48,132.26(2C),129.13(2C),127.69(2C),125.25(2C),124.30,117.94,115.99,113.61,111.70,104.46,73.37,63.74,57.81(2C),54.24(2C).
HMRS-ESI:m/z[M+H] calcd. for C29H27F2N6O: 529.2164; found: 529.2158.
③ {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(呋喃甲酰-2-基)-1-哌嗪基]}-2-丙醇(7d)的合成
紫色蜡状固体化合物,产率为48.5%.
1H NMR (400 MHz,CDCl3) δ 8.18 (s,1H),7.93 (s,1H),7.60 (s,1H),7.51 (s,4H),7.45~7.42 (m,1H),7.03~6.87 (m,3H),6.45 (dd,J=3.4,1.7 Hz,1H),4.65 (s,1H),4.49 (d,J=14.4 Hz,2H),4.40 (d,J=14.4 Hz,2H),3.71 (s,4H),3.08 (d,J=13.4 Hz,1H),2.83 (d,J=13.4 Hz,1H),2.48 (d,J=5.5 Hz,2H),2.40 (s,2H).
13C NMR (101 MHz,CDCl3) δ 165.87(C=O),163.70,161.44,158.97,150.92,147.60,144.88,143.77,142.57,134.50,131.32,129.16(2C),125.26 (2C),124.48,118.14,111.88,111.34,104.46,73.13,63.67,58.09 (2C),54.44 (2C).
HMRS-ESI:m/z[M+H] calcd. for C26H26F2N5O3: 494.2004; found: 494.2009.
④ {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(噻吩甲酰-2-基)-1-哌嗪基]}-2-丙醇(7e)的合成
紫红色蜡状固体化合物,产率为46.5%.
1H NMR (400 MHz,CDCl3) δ 8.13 (s,1H),7.90 (s,1H),7.51 (s,4H),7.41 (td,J=8.8,5.7 Hz,2H),7.24~7.18 (m,1H),7.00 (dd,J=4.9,3.7 Hz,1H),6.94 (dd,J=9.3,6.7 Hz,2H),4.64 (s,1H),4.48 (d,J=14.4 Hz,1H),4.37 (d,J=14.4 Hz,1H),3.62 (s,4H),3.17 (s,1H),3.05 (d,J=13.4 Hz,1H),2.78 (d,J=13.4 Hz,1H),2.47 - 2.39 (m,2H),2.34 (d,J=4.5 Hz,2H).
13C NMR (101 MHz,CDCl3) δ 166.78( C=O ),163.89,161.26,158.47,150.68,144.85,143.87,142.67,134.52,131.35,130.82,130.65,130.18,129.18(2C),128.12,125.26 (2C),124.48,111.76,104.64,73.21,63.69,58.12 (2C),54.35 (2C).
HMRS-ESI:m/z[M+H] calcd. for C26H26F2N5O2S: 510.5787; found: 510.5794.
⑤ {1-(1H-1,2,4-三氮唑-1-基)-2-(2′,4′-二氟联苯-4-基)-3-[4-(环丙甲酰基)-1-哌嗪基]}-2-丙醇(7f)的合成
黄色蜡状化合物,产率为62.9%.
1H NMR (400 MHz,CDCl3) δ 8.11 (s,1H),7.88 (s,1H),7.50 (s,4H),7.40 (td,J=8.6,6.6 Hz,1H),7.04 - 6.80 (m,2H),4.47 (d,J=14.3 Hz,1H),4.37 (d,J=14.3 Hz,1H),3.79 (s,1H),3.53 (s,4H),3.00 (d,J=13.4 Hz,1H),2.75 (d,J=13.4 Hz,1H),2.37 (s,2H),2.34~2.22 (m,2H),1.68~1.58 (m,1H),0.92 (dt,J=6.4,3.2 Hz,2H),0.71 (dd,J=7.8,3.0 Hz,2H).
13C NMR (101 MHz,CDCl3) δ 171.88 (C=O),163.64,161.08,158.52,151.20,144.86,142.87,134.40,131.32,129.09(2C),125.29 (2C),124.45,111.74,111.53,104.44,73.21,63.82 (2C),57.95 (2C),10.84,7.31 (2C).
HMRS-ESI:m/z[M+H] calcd. for C25H28F2N5O2: 468.2211; found: 468.2218.
1.2.2 目标化合物的体外抑菌试验 以氟康唑和酮康唑为阳性对照药物,以白色念珠菌(Candidaalbicans);新型隐球菌(Crytococcusneoformans);红色毛癣菌(Trichophytonrubrum)三种常见的人体治病真菌作为实验真菌菌株,参照美国国家临床试验室标准委员会推荐的标准化抗真菌敏感性实验方法[24],采用96孔微量稀释法测定目标化合物对各实验真菌的最低抑菌浓度(MIC80),既对实验真菌生长抑制率为80%时的最低抑菌浓度,实验结果见表 1.
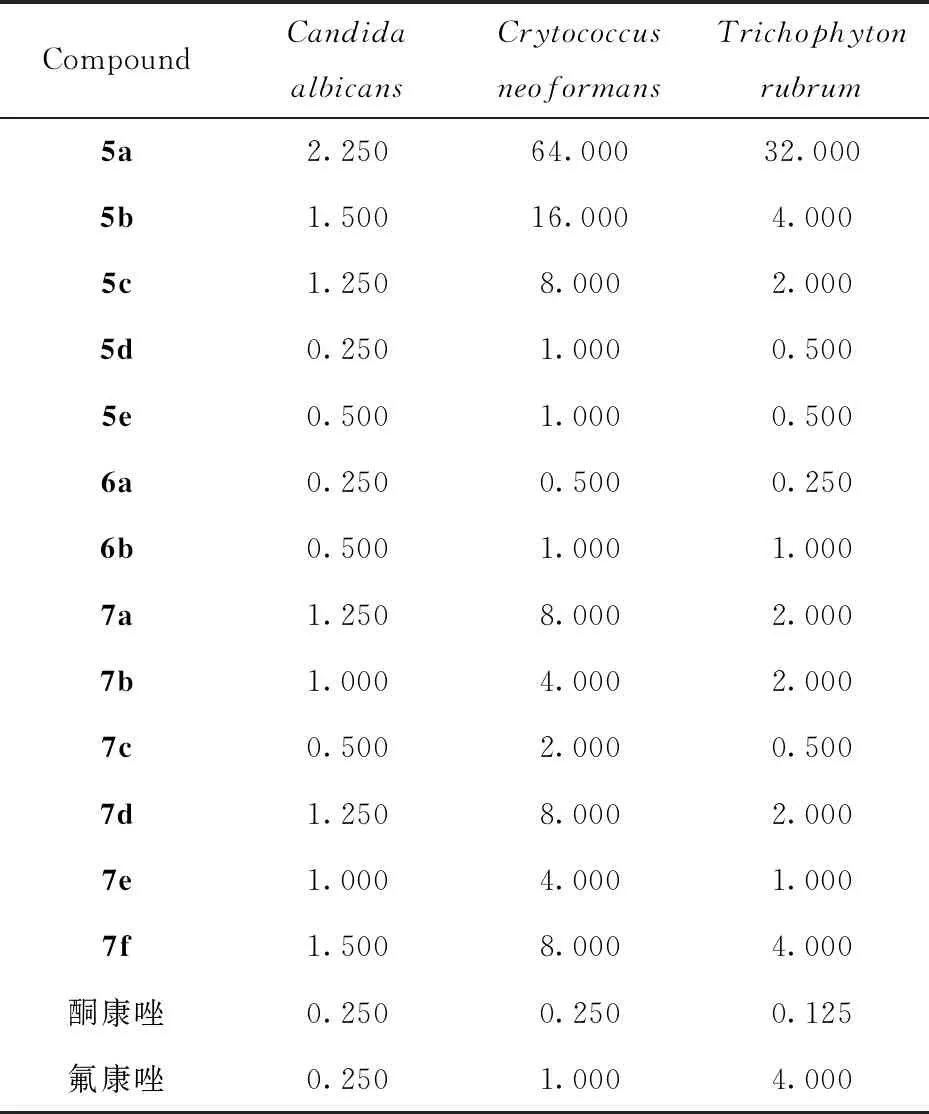
表1 目标化合物的体外抑菌活性Tab.1 In vitro antifungal activity (MIC80 ) of the target compounds /(μg·mL-1)
2 结果与讨论
2.1 标化合物合成条件优化
在目标化合物的合成中,中间体化合物4的合成是关键步骤,尝试了不同的反应条件对化合物4收率的影响,由表2可以看出,采用十六烷基三甲基溴化铵为相转移催化剂,甲苯为反应溶剂,经多次实验发现用20%NaOH溶液,反应温度60 ℃,反应时间1.5 h得到的反应结果最佳.
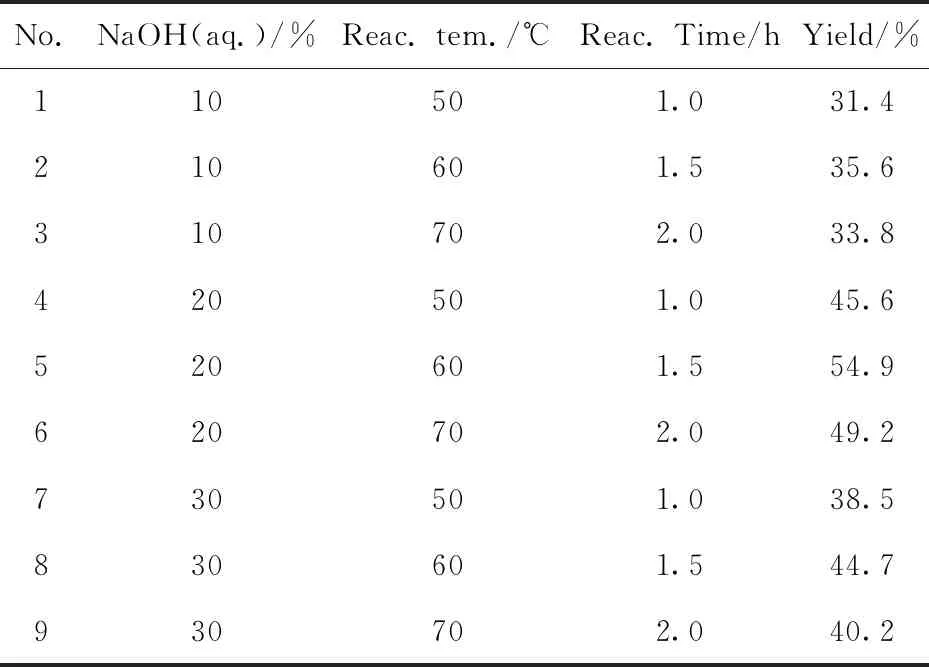
表2 不同反应条件对化合物4合成收率的影响Tab.2 Effect of reaction condition on the yield of the synthesis of compound 4
氢氧化钠溶液浓度过大、反应温度过高、反应时间过长将导致反应物中的—OH和生成的化合物4中的环氧乙烷基进一步反应,导致环氧乙烷开环,副产物增多,产物减少.化合物4和哌嗪反应可高产率得到化合物5a.化合物5a在多种溶剂中的溶解性较差,但作为中间体,在较多的二氯甲烷溶剂中和反应物酰氯经较长时间反应可得到目标产物6a,6b,7a~7f,目标产物在溶剂中溶解度较大,三乙胺在酰化反应中起到缚酸剂的作用,有利于产率的提高.
2.2 图谱分析
化合物4通过硅胶柱层析分离提纯后,得到的为棕红色粘稠状物质,所含溶剂很难出去,杂质峰较多,核磁图谱不能正确归属,但作为中间体不影响下一步反应.化合物5a经分离纯化后得到是白色固体产物,在CDCl3、d6-DMSO、D2O、CD3OD等溶剂中的溶解性极差,由于溶液浓度过低,核磁图谱测试没能成功.
目标化合物分子结构的相同之处是都含2,4-二氟联苯基、1,2,4-三氮唑环、哌嗪环、羟基以及和1,2,4-三氮唑环、哌嗪环直接相连的两个亚甲基,不同之处在于不同化合物分子分别接入了含有不同取代基的苯环或芳香杂环. 因此目标化合物的1H NMR和13C NMR图谱在相应的位置有相似或接近的化学位移值,但不同的化合物之间仍稍有差异.1H NMR图谱中,化学位移δ 8左右的两个单峰,分别为1,2,4-三氮唑环上的两个氢吸收峰;化学位移δ 7.5~6.5范围的多个吸收峰为2,4-二氟联苯基苯环上及分别接入目标化合物分子中含不同取代基的苯环或芳香杂环上的氢吸收峰,此处氢由于个数较多,且相互之间耦合,不同的氢吸收峰重合,峰形复杂,较难归属.—OH上氢的吸收峰为宽展的单峰,由于分子结构的差异,化学位移值有一定的差异,如:δ 4.84(5b,5c),δ 4.50(6a),δ 4.65(7d).和1,2,4-三氮唑环直接相连的亚甲基(—CH2—)上的氢吸收峰,化学位移值在δ 4.4左右,不同化合物之间稍有差异.和哌嗪环上的氮直接相连的亚甲基以及哌嗪环上的四个亚甲基上的氢吸收峰,化学位移值在大多在δ 3.45~2.32范围内,不同化合物之间稍有差异,吸收峰多有耦合分裂现象.化合物7f中在化学位移值δ 1.68~1.58 (m,1H),0.92 (dt,J=6.4,3.2 Hz,2H),0.71 (dd,J=7.8,3.0 Hz,2H) 处的吸收峰是环丙基上氢原子吸收峰.
13C NMR图谱中,和氟原子直接相连的苯环上的碳吸收峰,化学位移在δ 163,161.分子中其余芳香环上碳的吸收峰,化学位移在 δ 158~δ104范围内,不同芳香环上不同碳原子吸收峰,化学位移值 δ 有些相差很大,有些非常接近,有些不同碳原子化学位移值δ完全相同,吸收峰重合,如2,4-二氟联苯基不含氟原子的苯环上其中四个碳原子化学位移值 δ 129(2C),125 (2C).分子中亚甲基(—CH2—)上碳原子吸收峰,化学位移值在 δ 63~δ 45范围内,其中哌嗪环上四个碳的吸收峰,化学位移值 δ 54 (2C),48 (2C).和羟基直接相连的季碳吸收峰,化学位移值δ 75.化合物7f中在化学位移值δ 10.84,7.31 (2C) 处的吸收峰是环丙基上碳原子吸收峰.
2.3 目标化合物的抗真菌活性
体外抗真菌活性测试结果表明,大部分目标化合物对参与本实验的真菌生长有一定的抑制作用,其中目标化合物5d,5e,6a,6b,7c的抗真菌活性较好,化合物5d,6a对白色念珠菌的抑菌活性和酮康唑、氟康唑的抑菌活性相当,化合物5d,5e,6a,7c对红色毛癣菌的抑菌活性接近酮康唑,优于氟康唑.从构效关系看,化合物5b~5e抗真菌活性比较,和哌嗪环相连的嘧啶(5d)、吡啶(5e)杂环化合物的抗真菌活性优于相连基团为苯环的化合物(5b,5c).和哌嗪环相连的磺酰基类化合物(6a,6b)其抗真菌活性优于和哌嗪环相连的酰基类化合物(7a~7f).
3 结论
依据药物分子设计原理,设计合成了以 2,4-二氟代联苯基为分子骨架的新型三氮唑化合物.研究了各步合成反应的最佳反应条件.分析了目标化合物的核磁图谱,其分子结构经核磁共振氢谱、碳谱、高分辨质谱进行确证.测试了目标化合物的体外抗真菌活性,测试结果表明,大部分目标化合物对参与本研究的真菌生长有抑制一定的作用,其中目标化合物5d,5e,6a,6b,7c的抗真菌活性较好,化合物5d,6a对白色念珠菌的抑菌活性和酮康唑、氟康唑的抑菌活性相当,化合物5d,5e,6a,7c对红色毛癣菌的抑菌活性接近酮康唑,优于氟康唑.
猜你喜欢
分子催化(2022年1期)2022-11-02
石油化工(2022年9期)2022-10-19
中老年保健(2022年3期)2022-08-24
城市道桥与防洪(2022年3期)2022-05-08
快乐学习报·教师周刊(2022年3期)2022-04-21
化学工业与工程(2022年1期)2022-03-29
安全与环境工程(2021年2期)2021-04-02
化工设计通讯(2021年4期)2021-01-07
煤炭加工与综合利用(2020年6期)2020-07-17
农产品加工(2019年19期)2019-10-23
