
熵权TOPSIS法结合多指标成分综合评价金钱草药材的质量
2020-12-28 11:52严玉晶崔婷丁青洪婉敏李秀枝孙冬梅
中国药房 2020年23期
关键词:含量测定
严玉晶 崔婷 丁青 洪婉敏 李秀枝 孙冬梅

摘 要 目的:建立金錢草药材的超高效液相色谱(UPLC)特征指纹图谱,同时测定其中3种有效成分的含量,并综合评价不同产地金钱草的整体质量。方法:采用UPLC法建立10批金钱草全草、茎及叶的特征指纹图谱并测定山柰酚-3-O-芸香糖苷、槲皮素、山柰素的含量。色谱柱为Waters CORTECS UPLC T3,流动相为乙腈-0.2%磷酸水溶液(梯度洗脱),检测波长为364 nm,柱温为30 ℃,流速为0.2 mL/min,进样量为1 ?L。采用《中药色谱特征图谱相似度评价系统(2012版)》进行相似度评价,确定共有峰;以山柰酚-3-O-芸香糖苷、槲皮素、山柰素、总灰分、酸不溶性灰分、二氧化硫残留量及醇溶性浸出物为指标,采用熵权优劣解距离法(TOPSIS)对金钱草药材的整体质量进行综合评价。结果:10批金钱草全草、茎及叶共有7个共有峰,指认了其中3个峰分别为山柰酚-3-O-芸香糖苷、槲皮素、山柰素。同一部位不同批次金钱草全草的相似度均不低于0.830;同一批次金钱草茎与叶的相似度为0.504~0.859,全草与茎的相似度为0.593~0.904,全草与叶的相似度为0.885~0.995。山柰酚-3-O-芸香糖苷、槲皮素、山柰素检测质量浓度的线性范围分别为0.392 0~39.197 0、0.397 0~39.703 4、0.380 9~38.093 0 μg/mL(r均大于0.999 0);精密度、稳定性、重复性试验的RSD均小于2%;加样回收率分别为96.43%(RSD=0.63%,n=9)、100.32%(RSD=0.46%,n=9)、101.80%(RSD=0.32%,n=9)。金钱草中上述3种成分的含量分别为0.006 3%~0.041 1%、0.002 9%~0.008 6%、0.004 4%~0.017 5%(茎), 0.024 8%~0.290 5%、0.000 9%~0.009 0%、0.001 3%~0.012 4%(叶),0.007 9%~0.118 0%、0.001 5%~0.008 8%、0.002 8%~0.012 5%(全草)。不同产地样品间3种成分含量比较,差异均无统计学意义(P>0.05);金钱草不同部位中山柰酚-3-O-芸香糖的含量大小顺序依次为叶>全草>茎,槲皮素与山柰素的含量大多以茎中含量较高。熵权TOPSIS法结果显示,四川省中江县、双流县以及重庆市石柱县产金钱草的最优解欧氏贴近度均值分别为0.446、0.512、0.287。结论:所建特征指纹图谱和含量测定方法稳定、可行,结合熵权TOPSIS法所建的多指标评价模型可用于金钱草药材质量的综合评价;四川省产金钱草药材质量较优。
关键词 金钱草;不同部位;超高效液相色谱法;含量测定;特征指纹图谱;熵权优劣解距离法
ABSTRACT OBJECTIVE: To establish UPLC characteristics fingerprints of Lysimachia christinae, and to simultaneously determine 3 effective components and to comprehensively evaluate the quality of L. christinae from different production areas. METHODS: UPLC method was adopted to establish characteristics fingerprint of the whole plant, stem and leaves of 10 batches of L. christinae, and determine the contents of kaemperfol-3-O-rutinoside, quercetin, kaemperfol. The determination was performed on Waters CORTECS UPLC T3 column with mobile phase consisted of acetonitrile-0.2% phosphoric acid aqueous solution (gradient elution) at the flow rate of 0.2 mL/min. The detection wavelength was set at 364 nm, and column temperature was 30 ℃. The sample size was 1 ?L. Similarity Evaluation System for TCM Chromatographic Fingerprint(2012 edition) was adopted to evaluate its similarity, and common peaks were confirmed. Using the contents of kaemperfol-3-O-rutinoside, quercetin, kaemperfol, total ash, acid-insoluble ash and sulfur dioxide residue, the ethanol-soluble extract as index, entropy weight TOPSIS was used to evaluate the overall quality of L. christinae comprehensively. RESULTS: There were 7 common peaks in the whole plant, stem and leaves of 10 batches of L. christinae, among which 3 peaks were identified as kaemperfol-3-O-rutinoside, quercetin and kaemperfol. The similarity of same part in the whole plant of L. christinae from different batches were not lower than 0.830. The similarity between stem and leaves of L. christinae in same batch was 0.504-0.859; the similarity between whole plant and stem was 0.593-0.904; the similarity between whole plant and leaves was 0.885-0.995. The linear ranges were 0.392 0-39.197 0 μg/mL for kaempferol-3-O-rutinoside, 0.397 0- 39.703 4 μg/mL for quercetin, 0.380 9-38.093 0 μg/mL for kaempferol (r>0.999 0). RSDs of precision, stability and repeatability tests were all lower than 2%. The recoveries were 96.43% (RSD=0.63%, n=9), 100.32% (RSD=0.46%, n=9), 101.80% (RSD=0.32%, n=9) ,respectively. The content range of above components in L. christinae were 0.006 3%-0.041 1%, 0.002 9%-0.008 6%, 0.004 4%-0.017 5%(stem); 0.024 8%-0.290 5%, 0.000 9%-0.009 0%, 0.001 3%-0.012 4%(leaves); 0.007 9%-0.118 0%, 0.001 5%-0.008 8%, 0.002 8%-0.012 5%(whole plant). There was no significant difference in the contents of 3 components in L. christinae among different producing areas (P>0.05). The order of the contents of kaempferol-3-O- rutinoside in different parts of L. christinae was leaves>whole plant>stem. The contents of quercetin and kaempferol were high relatively in the stem. Results of entropy weight TOPSIS method showed that mean values of Ci for L. christinae from Zhongjiang county and Shuangliu county of Sichuan province, Shizhu county of Chongqing city were 0.446,0.512,0.287. CONCLUSIONS: Established fingerprint and content determination method are stable and feasible, and multi-index evaluation model constructed by characteristic chromatogram combined with entropy weight TOPSIS analysis method can be used for comprehensive quality evaluation of L. christinae. The quality of L. christinae from Sichuan province is better.
2.1.5 稳定性试验 取“2.1.3”项下供试品溶液(编号:S1)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.1.1”项下色谱条件进样测定,以槲皮素为参照,计算各共有峰的相对保留时间和相对峰面积。结果,7个共有峰相对保留时间的RSD≤0.48%(n=6),相对峰面积的RSD≤1.14%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.1.6 重复性试验 取金钱草样品粉末(编号:S1)约1.0 g,精密称定,共6份,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,以槲皮素为参照,计算各共有峰的相对保留时间和相对峰面积。结果,7个共有峰相对保留时间的RSD≤0.49%(n=6),相对峰面积的RSD≤1.98%(n=6),表明方法重复性良好。
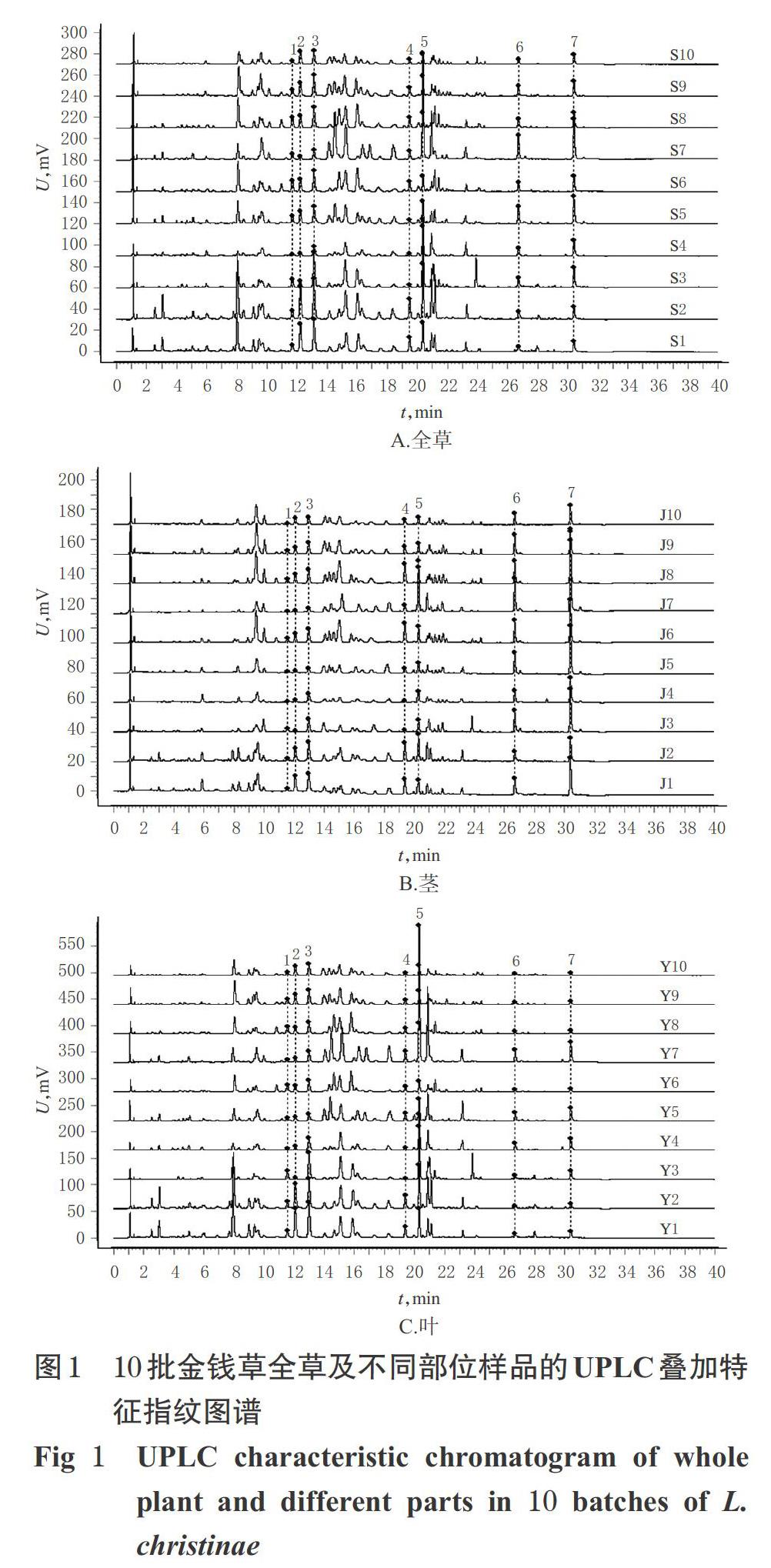
2.1.7 特征指纹图谱的建立 取金钱草全草及不同部位样品各10批,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,记录色谱图,采用《中药色谱指纹图谱相似度评价系统(2012版)》建立特征指纹图谱。分别以S1、J1、Y1样品色谱图为参照图谱,设置时间窗宽度为0.1 min,通过多点校正、全谱峰匹配分别生成共有特征指纹图谱,同时采用中位数法分别生成对照特征指纹图谱。结果,每批金钱草全草及不同部位样品均有7个共有峰,不同批次的金钱草相同部位间的色谱峰相似,化学成分整体差异较小,详见图1。
2.1.8 共有峰的指认 取“2.1.2”项下混合对照品溶液适量,按“2.1.1”项下色谱条件进样测定,记录色谱图。通过与混合对照品图谱比对,共指认3个共有峰,分别为山柰酚-3-O-芸香糖苷(5号峰)、槲皮素(6号峰)、山柰素(7号峰)。因6号峰槲皮素的分离度较优、峰形较好,故以其为参照,计算其他各共有峰的相对保留时间和相对峰面积,详见图2。
2.1.9 相似度评价 采用《中药色谱指纹图谱相似度评价系统(2012版)》进行相似度评价,结果见表2、表3。由图1、表2可知,同一部位不同批次金钱草的特征图谱基本相似,与其对照特征指纹图谱一致性较好,相似度均不低于0.830,表明同一部位各批次间金钱草的化学成分差异较小。由表3可知,同一批次金钱草茎与叶的相似度为0.504~0.859,全草与茎的相似度为0.593~0.904,全草与叶的相似度为0.885~0.995,表明同一批次不同部位间金钱草的化学成分含量存在较大差异,其中金钱草全草与茎的相似度较低,而全草与叶的相似度较高,提示叶的化学成分与全草相似。
2.2 含量测定
2.2.1 色谱条件 同“2.1.1”项。
2.2.2 溶液的制备 混合对照品溶液的制备同“2.1.2”项,供试品溶液的制备同“2.1.3”项。取80%甲醇溶液,经0.22 ?m 微孔滤膜滤过,取续滤液,即得阴性对照溶液。
2.2.3 系统适用性试验 取“2.2.2”项下混合对照品溶液、供试品溶液、阴性对照溶液各适量,按“2.2.1”项下色谱条件进样测定,记录色谱图。结果,各待测成分与相邻色谱峰的分离度均大于1.5,理论板数按山柰酚-3-O-芸香糖苷、槲皮素、山柰素计均大于200 000,且阴性对照无干扰,详见图3。
2.2.4 线性关系考察 取“2.2.2”项下混合对照品溶液各0.1、0.5、1、2.5、5、10 mL,分别置于10 mL量瓶中,加80%甲醇稀释并定容,摇匀,得系列线性工作溶液。取上述工作溶液适量,按“2.2.1”项下色谱条件进样测定,记录色谱图。以各待测成分质量浓度为横坐标(X,μg/mL)、峰面积(Y)为纵坐标进行线性回归。结果,山柰酚-3-O-芸香糖苷的回归方程为Y=5 360.4X-70.392(r=0.999 9),槲皮素的回归方程为Y=18 957.0X-8 616.700(r=0.999 8),山柰素的回归方程为Y=23 319.0X-10 425.000(r=0.999 9),表明上述3种成分检测质量浓度的线性范围分别为0.392 0~39.197 0、0.397 0~39.703 4、0.380 9~38.093 0 μg/mL。
2.2.5 精密度试验 精密吸取“2.2.4”项下线性工作溶液(山柰酚-3-O-芸香糖苷、槲皮素、山柰素的质量浓度分别为26.131 3、26.468 9、25.395 3 μg/mL),按“2.2.2”项下色谱条件连续进样测定6次,记录峰面积。结果,山柰酚-3-O-芸香糖苷、槲皮素、山柰素峰面积的RSD分别为0.82%、0.51%、0.70%(n=6),表明仪器精密度良好。
2.2.6 稳定性试验 精密吸取“2.2.2”项下供试品溶液(编号:S1)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,山柰酚-3-O-芸香糖苷、槲皮素、山柰素峰面积的RSD分别为1.11%、0.99%、1.02%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.2.7 重复性试验 精密称取金钱草样品粉末(编号:S1),约1.0 g,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品含量。结果,山柰酚-3-O-芸香糖苷、槲皮素、山柰素的平均含量分别为0.033 6%、0.001 9%、0.003 4%,RSD分別为1.52%、1.43%、1.60%(n=6),表明方法重复性良好。
2.2.8 加样回收率试验 精密称取已知含量(含山柰酚-3-O-芸香糖苷0.033 6%、槲皮素0.001 9%、山柰素0.003 4%)的金钱草样品粉末(编号:S1),约0.5 g,共9份,按对照品与供试品中指标成分质量之比0.5 ∶ 1、1 ∶ 1、1.5 ∶ 1,分别加入一定量的混合对照品溶液,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果,3个成分的平均加样回收率分别为96.43%(RSD=0.63%,n=9)、100.32%(RSD=0.46%,n=9)、101.80%(RSD=0.32%,n=9)。
2.2.9 样品含量测定 取金钱草全草及不同部位样品粉末各10批,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品含量(按干燥品计)。每批样品平行操作2次。采用SPSS 26.0软件对数据进行统计分析,数据均以x±s表示,组间比较采用单因素方差分析;P<0.05为差异有统计学意义。样品含量测定结果见表4、表5。
由表5可知,不同产地金钱草全草及不同部位样品中3种成分含量分别比较差异均无统计学意义(P>0.05),表明不同产地金钱草整体质量较稳定;不同部位金钱草中山柰酚-3-O-芸香糖的含量大小顺序依次为叶>全草>茎;槲皮素与山柰素的含量大多以茎中含量较高。这提示,金钱草的茎为槲皮素和山柰素的富集部位,而叶则是山柰酚-3-O-芸香糖的富集部位。
2.3 熵权TOPSIS法分析
2.3.1 归一化处理 取10批金钱草药材(全草),分别按照2015年版《中国药典》(四部)通则“2302灰分测定法”[19]测定总灰分、酸不溶性灰分,按照通则“2331二氧化硫残留量测定法”第一法[19]测定二氧化硫残留量,按照通则“2201浸出物测定法”热浸法[19]测定醇溶性浸出物(用75%乙醇作溶剂)。同时,根据含量测定结果(表4),按公式(1)(2)对原始试验数据进行归一化处理[16-17]:
式中,Rij表示不同指标类型;Xij表示多目标决策矩阵,其中i表示样本值(i=1,2,……m),j表示指标值(j=1,2,……m) 。各指标归一化处理结果见表6。
2.3.2 加权决策矩阵 熵权法是基于评价指标的信息熵大小与差异离散程度,计算获得各指标权重(Wj),以客观反映各批金钱草样品在综合质量评价过程中的重要性[16-17]。金钱草中山柰酚-3-O-芸香糖苷、槲皮素、山柰素、总灰分、酸不溶性灰分、二氧化硫残留量及醇溶性浸出物的权重(Wj)=(0.259,0.155,0.131,0.073,0.079,0.117,0.186)。计算方法见公式(3)[16-17]:
式中,Z表示加权决策矩阵,加权决策矩阵结果见表7。
2.3.3 最优与最劣方案的确定 根据加权决策矩阵得到最优方案[Z +,Z +=max(Z1,Z2,…Zm) ]和最劣方案[Z-,Z-=min(Z1,Z2,…Zm)] [16-17]。结果,Z +=(0.259,0.155,0.131,0.073,0.079,0.117,0.186),Z-=(0,0,0,0,0,0,0)。
2.3.4 欧氏贴近度的计算及质量评价 根据最优与最劣方案,按公式(4)(5)(6)计算金钱草样品与最优方案的距离(D+)、与最劣方案的距离(D-)及最优解的欧氏贴近度(Ci)[16-17]:
D+越小、D-越大、Ci越大,则被评价样品越优[16-17]。金钱草质量评价结果见表8。
由表8可知,Ci排名前3位的依次為S3、S5、S7,其中1批(S3)来自四川省中江县、2批(S5、S7)来自四川省双流县。四川省中江县、双流县以及重庆市石柱县产区的Ci均值依次为0.446、0.512、0.287,表明四川产金钱草质量优于重庆产样品。
3 讨论
通过查阅文献发现,金钱草的特征图谱检测波长主要集中在240 nm和360 nm 左右[20-23],故本研究对240、310、364 nm波长进行考察。结果发现,以240、310 nm为检测波长时,各色谱峰响应值相差较大且基线不平稳;而当以364 nm为检测波长时,各色谱峰信号响应值较高且色谱峰分离度较好、基线较平稳,故选择364 nm为检测波长。同时,本研究又对不同提取方式(超声、回流)、提取溶剂(80%甲醇、50%甲醇、甲醇、无水乙醇、稀乙醇等)和提取时间(10、20、30 min)进行了考察。结果发现,以80%甲醇、超声处理20 min时,各指标成分含量较高,共有峰峰形较好,色谱峰分离度均大于1.5,故采用“2.1.3”项下制备方法。
为客观评价金钱草药材质量,本研究建立了不同产地金钱草全草及不同部位的特征指纹图谱,并对其中山柰酚-3-O-芸香糖苷、槲皮素、山柰素的含量进行了比较。结果显示,10批金钱草全草及不同部位样品均有7个共有峰,指认了山柰酚-3-O-芸香糖苷、槲皮素、山柰素3个共有峰;金钱草茎与叶相似度较低,全草与茎的相似度较低、与叶的相似度较高。不同产地金钱草全草及不同部位的3种成分含量在叶、全草中差异较小,在茎中差异较大,提示叶的化学成分与全草相似、含量接近。叶为山柰酚-3-O-芸香糖的富集部位,茎为槲皮素和山柰素的富集部位,故为保证疗效的一致性,在储存该药材时,应注意药材的完整性。熵权TOPSIS结果显示,四川省双流县产金钱草药材的Ci均值高于其余2个产地样品,提示四川省双流县产金钱草药材的整体质量较佳。
本研究所建特征指纹图谱与含量测定方法稳定、可行,可用于评价和比较金钱草药材的整体质量及茎、叶的化学成分差异,明确有效成分的富集部位,为进一步探讨金钱草不同部位化学成分引起的药效差异及药材资源的合理利用提供了数据支撑,也为该药材的道地性研究提供了基础,进一步促进了金钱草优质种源的研究与开发。
参考文献
[ 1 ] 国家药典委员会.中华人民共和国药典:一部[S]. 2015年版.北京:中国医药科技出版社,2015:219-220.
[ 2 ] 国家中医药管理局《中华本草》编委会.中华本草:第6册[M].上海:上海科学技术出版社,1999:94-95.
[ 3 ] 刘隽,邹国林.金钱草的研究进展[J].唐山师范学院学报,2002,24(2):8-10.
[ 4 ] 高飞飞.金钱草的化学成分研究[D].重庆:西南大学,2013.
[ 5 ] 汪丹,蔡甜,吴志军,等.液质联用分析金钱草醇提物中的化学成分[J].成都大学学报(自然科学版),2015,34(3):234-237.
[ 6 ] 陶婷婷,赵凡,叶妙勇,等.金钱草总黄酮对草酸钙结石模型大鼠肾组织骨桥蛋白表达的影响[J].浙江中西医结合杂志,2019,29(8):623-626.
[ 7 ] 王洋.金钱草活性成分及药理作用研究[D].西宁:青海师范大学,2018.
[ 8 ] 王洋,段培琪,王华清,等.小叶金钱草醇提物的抗炎、镇痛及利胆作用研究[J].华西药学杂志,2018,33(3):267-269.
[ 9 ] 吴敦锋.复方金钱草颗粒对肾结石患者利尿、解痉、抗炎作用效果探究[J].北方药学,2016,13(10):77-78.
[10] 吴学敏,顾成娟,欧文娥,等.大叶金钱草、鸡内金、桃仁治疗尿石症经验:仝小林三味小方撷萃[J].吉林中医药,2020,40(7):868-870.
[11] 李娜.红豆杉药效物质基础研究及中药金钱草质量评价模式探究[D].重庆:重庆医科大学,2018.
[12] 李可.四川省不同来源地金钱草种质资源的遗传多样性研究[D].雅安:四川农业大学,2009.
[13] 李清林,陶锋.浙江省不同产地金钱草总黄酮含量的测定[J].中国中医药科技,2017,24(1):49-50.
[14] 王俊文,熊颖,陈晓虎,等.过路黄和广金钱草的HPLC指纹图谱比较研究[J].中草药,2016,47(5):827-831.
[15] 文晶晶,黃桂红,韦奕,等.广金钱草和金钱草不同部位总黄酮含量研究[J].中国民族民间医药,2019,28(8):58- 60.
[16] 李运,张霁,徐福荣,等.多指标决策分析TOPSIS对三七质量评价研究[J].中草药,2017,48(22):4764-4771.
[17] 马天翔,顾志荣,许爱霞,等.基于OPLS结合熵权TOPSIS法对不同产地锁阳的鉴别与综合质量评价[J].中草药,2020,51(12):3284-3291.
[18] 陈涛,胡君,吕明远,等.基于超高效液相色谱法(UPLC)测定芪丹利心丸中丹酚酸B的含量[J].山西中医,2018,34(11):50-53.
[19] 国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:202、204、208.
[20] 周祎,郜红利.HPLC-DAD法同时测定中药金钱草中6种活性成分的含量[J].中国药师,2016,19(8):1609-1611.
[21] 郭林林,赵德,邓君.金钱草药材的HPLC指纹图谱研究[J].中药材,2012,35(3):382-385.
[22] 金丽鑫.金钱草对照药材标定技术标准的研究[D].成都:成都中医药大学,2012.
[23] 方国花.金钱草超微粉、总黄酮富集及药材指纹图谱研究[D].吉林:长春中医药大学,2010.
(收稿日期:2020-09-09 修回日期:2020-11-04)
(编辑:陈 宏)
猜你喜欢
考试周刊(2016年103期)2017-01-23
中国民族民间医药·下半月(2016年11期)2017-01-19
中国民族民间医药·下半月(2016年12期)2017-01-19
中国医药导报(2016年28期)2017-01-06
中国医药导报(2016年30期)2016-12-28
云南中医中药杂志(2016年11期)2016-12-26
中国民族民间医药·上半月(2016年11期)2016-12-26
中国民族民间医药·上半月(2016年11期)2016-12-26
考试周刊(2016年95期)2016-12-21
