
基于3D-QSAR和分子对接设计新型缺氧诱导因子脯氨酸羟化酶结构域蛋白1抑制剂
2020-12-24 07:52:52何华玉何清秀林治华
重庆理工大学学报(自然科学) 2020年11期
储 涵,何华玉,何清秀,王 娟,林治华
缺氧诱导因子(HIFs)是由氧气敏感的α亚基(HIF-1α、HIF-2α或 HIF-3α)和组成性表达的 β亚基(HIF-1β、HIF-2β或 HIF-3β)组成的异源二聚体转录因子[1]。在常氧环境中,HIF-α的羟基化和随后的蛋白酶体降解能够通过泛素连接酶系统对HIFs进行负调控[2-3]。同时,HIFs也是有限氧环境中氧稳态的主要调节器[4]。有研究表明:HIFs的稳定性主要由HIF-α的翻译后脯氨酰羟基化来调节,该反应由缺氧诱导因子脯氨酸羟化酶结构域蛋白(PHDs)催化[5-6]。PHDs是一组在多细胞动物中充当氧气感受器的酶,同时PHDs作为一种非血红素铁(II)依赖的酶,有3种催化活性亚型(PHD-1,PHD-2和 PHD-3)[7-8]。其中,PHD-1主要存在于睾丸中,但也存在于脑、肾、心脏和肝脏中,PHD-2存在于大多数组织中,PHD-3只存在于心脏中[9-10]。这3种异构体在其C端催化结构域具有很高的序列同源性,但在其N端没有序列同源性[11]。
目前,在诸多临床环境中,包括炎症性肠病,缺血再灌注损伤和神经退变过程中,干扰PHDHIF轴成为了一种重要的治疗方式[12-17]。常规的干扰方式(如Fe2+隔离剂等),由于缺乏特异性,在使用过程中存在很强的副作用[18-19]。1,2,4-三唑-[1,5-a]吡啶类化合物作为一类新型特异性PHD-1抑制剂,通过靶向 PHD-1干扰 PHD-HIF轴,避免了非特异抑制剂的副作用,也大大增强了疗效。在本研究中,课题组收集了44个1,2,4-三唑-[1,5-a]吡啶类化合物,通过比较分子场分析(CoMFA)和比较分子相似性指数分析(CoMSIA),获得了可靠的3D-QSAR模型。随后,通过分子对接进一步分析了PHD-1与该类化合物的结合模式。最后,基于上述结果,设计了8种新型1,2,4-三唑-[1,5-a]吡啶类 PHD-1抑制剂,并确定可用于进一步研究的先导化合物。
1 材料与方法
1.1 数据选取
首先,根据文献收集了 44个 1,2,4-三唑-[1,5-a]吡啶类化合物[20],将它们随机分为训练(36种化合物)和测试数据集(8种化合物,用符号“*”表示)。这些化合物的体外生物活性值以IC50表示,并转换为相应的 pIC50(pIC50=-log IC50)。化合物的相关属性见表1。
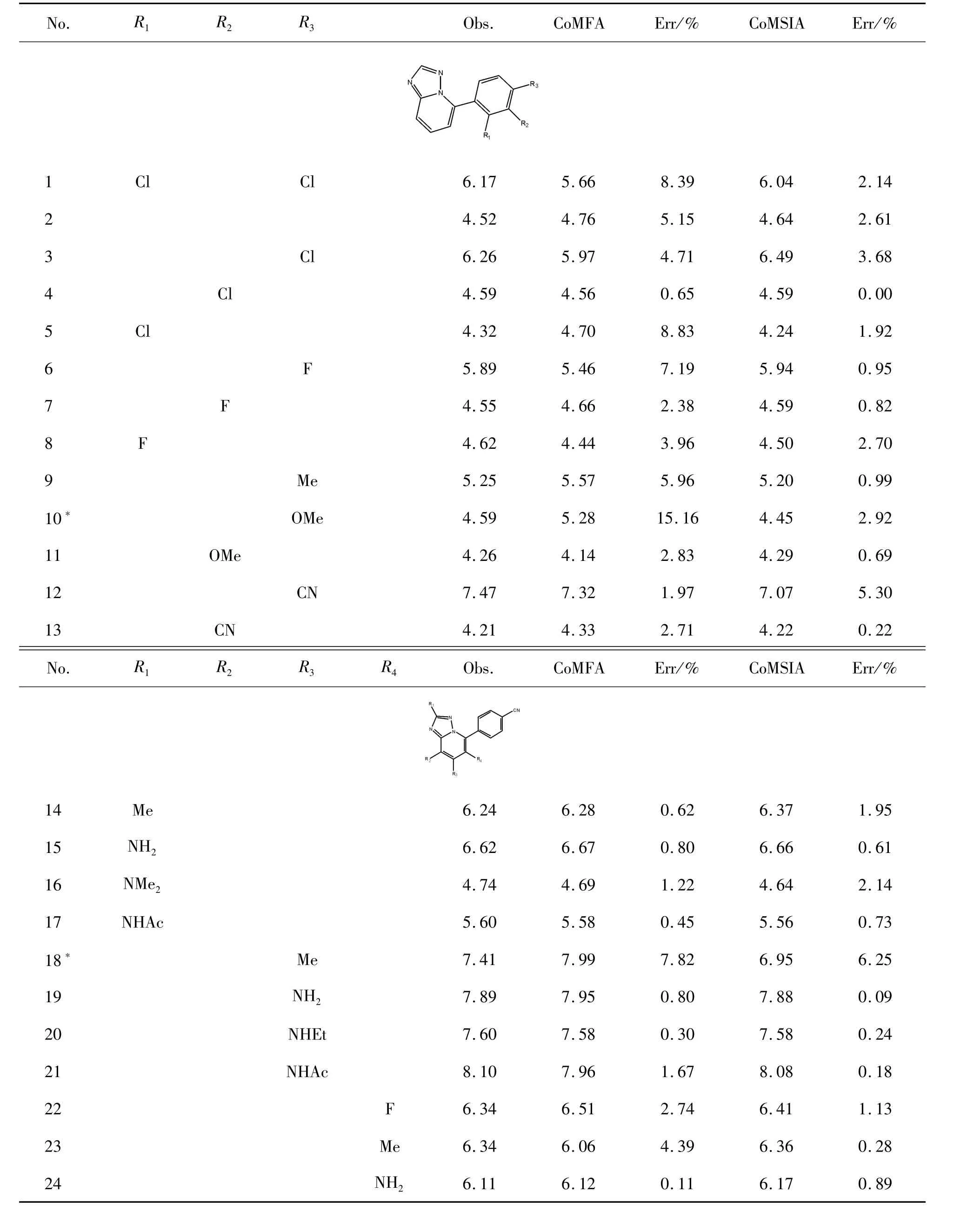
表1 化合物的实验和预测活性(pIC50)值

续表(表1)
1.2 结构优化和叠合
利用sybyl 2.0构建这44个化合物,然后给每个化合物附加Gasteiger-Huckel电荷和Tripos力场。采用Powll能量梯度法对化合物的结构进行优化,最大优化上限10 000次,收敛标准为0.005 kJ/mol,其余参数默认[21]。所有处理过后的化合物构象作为活性构象用于下一步的研究,同时化合物21(如图1,图中加粗部分表示公共骨架)具有最高的抑制活性,可作为叠合模板。选择适当的公共骨架后,所有化合物的叠合如图2所示。
1.3 构建3D-QSAR模型
CoMFA通过分析一组相似化合物周围分子场的差异,得到这些分子与受体之间的非间相互作用的特性,从而预测其生物活性。CoMFA的静电场和立体场分别采用Coalomb和Lennard-Jones势函数,而CoMSIA还引入了高斯距离函数,通过疏水,氢键受体和供体场计算探针与分子的每个原子之间的相似性指数[22-23]。通过留一法(LOO)交叉验证程序计算了相关模型的最佳组件数(n)和最高交叉验证相关系数(q2)。常规的多重相关系数(r2),估计的标准误差(SEE)和 Fisher检验(F)值是通过非交叉验证的分析获得的[24-25]。利用3D-QSAR模型的等势图研究分子结构对生物活性的影响,从而指导化合物结构的修饰以提高其活性。
1.4 分子对接
PHD-1(PDB∶5v1b)晶体结构来源于蛋白数据库(protein data bank,PDB),利用 Sybyl软件对PHD-1晶体结构进行加氢、加电荷等处理,同时,去除晶体结构中的原配体小分子(8UY)。将原配体小分子范围内0.5 nm的氨基酸残基作为对接口袋区域,并以其为参考分子,其余参数为默认值[26]。基于上述参数,将所有化合物与其对接,如图3,使用PyMOL软件进行对接可视化。
2 结果与讨论
2.1 3Q-QSAR模型
以36个 1,2,4-三唑-[1,5-a]吡啶类化合物为训练集,CoMFA和CoMSIA建模后的计算结果显示在表2中。CoMFA模型和CoMSIA模型的稳定性可以用交叉验证的q2来表示。一般情况下,当q2大于0.3时,所构建的模型仅在5%水平上有统计学意义;当q2大于0.5时,模型具有显著的统计学意义。
空间场和静电场构建的CoMFA模型最佳,LOO的q2为0.712,表明该模型具有良好的稳定性和可预测性。此外,CoMFA模型的n值为7,r2为0.969,F值为123(认为可靠的F>100),SEE为0.222,说明该模型具有良好的统计意义。在CoMSIA模型中,空间场、静电场、疏水场,氢键供体场和受体场的组合表现良好(q2=0.754,r2=0.985),同时其余参数也可证明(F=159,SEE=0.165)。3D-QSAR模型的训练集与测试集的线性相关性分析如图4所示。CoMFA与CoMSIA模型中训练集与测试集的大部分化合物都位于或接近趋势线附近,证明了化合物实际活性值与预测活性值(以pIC50表示)的拟合度较好。

表2 3D-QSAR模型的计算结果
2.2 CoMFA和CoMSIA三维等势图分析
使用实际活性最高的的化合物21作为模板进行CoMFA与CoMSIA的三维等势图分析。如图5所示,CoMFA立体场、COMSIA静电场、疏水场和氢键受体场贡献较高,可用于指导化合物结构修饰和改造。
图5 (a)是 CoMFA的立体场轮廓图。绿色(贡献80%)和黄色(贡献20%)区域的组合表明:在化合物的苯环连接的-CN处以体积较大的基团取代能够增强活性。化合物10(pIC50值为4.59)在该处以较大体积的基团-OMe取代,相较于无取代基的化合物13(pIC50值为 4.21)活性更高。CoMSIA的静电场等势图见图5(b),蓝色区域表示在此处增加负电性基团有利于提高化合物的活性;红色区域表示在此处以正电性基团取代更好。这种结合表明:三唑环带有负电荷的基团有利于活性。化合物19(pIC50值为7.89)的-NH2取代负电性更强,比-CH3修饰的化合物18(pIC50值为7.41)有更优异的活性。CoMSIA的疏水场等势图见图5(c),白色区域表示在此处增加亲水性基团有利于提高化合物的活性,黄色区域表示在此处以疏水性集团取代更好。因此,在化合物的苯环连接的-CN处以亲脂性的基团取代能够增强活性。CoMSIA的氢键受体场等势图显示在图5(d)中。红色部分表示在此处减少氢键受体有利于活性的提升。因此,在苯环上的需要更多的非氢键供体。这些已通过系统中的某些化合物确认。在化合物26和27中,化合物27(pIC50值为7.30)以-H取代,提供氢键供体而非氢键受体,其活性明显高于-CF3取代的化合物26(pIC50值为5.73)。
2.3 分子对接模型分析
为了探索PHD-1与配体的结合方式,利用SYBYL软件的Surflex-Dock模块进行分子对接,如图6所示。在对接所有化合物前,将PHD-1共晶配体8UY重新对接到结合囊中来验证所使用的方法和参数是否可靠可行。如图7所示,8UY分子重新对接的构型与其共晶配体构型基本一致,两种构型的RMSD为0.2Å(<2.0Å)。化合物中的三唑环上的N原子直接与金属Fe结合,这种特征的相互作用与以往的研究是一致的。这也说明所生成的对接协议是可靠的,可用于后续的研究。
本研究选择化合物21作为分析结合模式的模板,并用二维图(图8)和三维图(图6)进行展示。首先,化合物21三唑环上的氮原子与活性部位的Fe形成Fe-N键(2.0Å)。与苯环相连的NH连接体与ASP238形成了一个距离为2.4Å的强氢键。值得注意的是化合物另一端的氰基与Asn315也形成了一个强氢键(2.3Å)。这种氢键的结合方式保证了其稳定性。此外,化合物21与PHD-1之间存在丰富的疏水相互作用。Tyr287与该化合物的苯环之间都存在Pi-Pi相互作用(3.3 Å)。此外,Ala369与该苯环之间也存在着一个疏水作用(5.9Å)。另外,Ile311与三唑环之间还存在着一个弱疏水作用(3.9Å)。丰富的疏水相互作用和氢键在配体与受体的结合中起着关键作用,它们可以使化合物与受体的结合更加稳定。
2.4 新型化合物的设计与评价
通过分析3D-QSAR的等势图和分子对接模型,以化合物21为先导化合物,确定该类化合物的修饰区域。通过在苯环附近引入氢键受体和体积较大的疏水性基团,在三唑环附近引入正电荷基团,本文共设计了 8个新的1,2,4-三唑-[1,5-a]吡啶类化合物。
对于这8个新设计的化合物,首先通过建立的3D-QSAR模型预测了其活性,其次将其与PHD-1对接并打分。化合物的结构,预测活性以及对接得分如表3所示。结果表明:所有新化合物抑制活性较高,且对接得分也较高,尤其是化合物21-g,其预测活性相较于化合物21有明显的提升,同时它与PHD-1的对接得分也同样有所提升。这表明,相较于其余修饰化合物的方式,在苯环附近引入氢键受体能够很好地提高该类化合物与PHD-1结合的稳定性,从而大大提升1,2,4-三唑-[1,5-a]吡啶类化合物作为特异性PHD-1抑制剂的活性。
同时,由于化合物21-g具有出色的预测活性和对接评分,因此它可作为新型 1,2,4-三唑-[1,5-a]吡啶类PHD-1抑制剂的先导化合物,用作进一步的研究。

表3 新设计化合物的结构和预测活性
3 结论
本文中结合3D-QSAR和分子对接技术研究了1,2,4-三唑-[1,5-a]吡啶类 PHD-1抑制剂。所建立的3D-QSAR模型揭示了PHD-1抑制剂结构与活性关系的关键因素,这些关键因素可用于指导该类化合物的改造和设计。此外,分子对接技术被用于探索蛋白晶体活性位点中的关键氨基酸与配体分子的对接模式。通过分析空间场、疏水场、静电场和氢键受体场的等势图,以及1,2,4-三唑-[1,5-a]吡啶类化合物与PHD-1的对接模型,设计了8种新的该类化合物。新型化合物评价结果表明:它们都具有良好的预测活性和较好的对接得分,尤其是化合物21-g,可作为先导化合物做进一步研究。这些结果为特异性PHD-1抑制剂的合理设计奠定了基础。
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
云南化工(2021年10期)2021-12-21 07:33:28
化工学报(2020年4期)2020-05-28 09:25:24
高中数理化(2020年1期)2020-02-29 02:21:18
今日农业(2019年11期)2019-08-13 00:49:02
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
中国果菜(2016年9期)2016-03-01 01:28:41
西华师范大学学报(自然科学版)(2015年3期)2015-02-27 15:31:19
火炸药学报(2014年5期)2014-03-20 13:17:47
无机化学学报(2014年7期)2014-02-28 17:32:28
