
无溶剂无催化合成异吲哚啉类化合物
2020-12-23 13:00:42钟凯凯刘金彪谢志强卢俊瑞
天津理工大学学报 2020年6期
钟凯凯,刘金彪,谢志强,卢俊瑞
(1.天津理工大学化学化工学院,天津300384;2.天津瑞岭化工有限公司,天津300384)
异吲哚啉类化合物是一类具有广泛的生物活性的生物碱,研究发现有许多含有异吲哚啉骨架环结构的化合物有重要的药理作用,在医药领域的开发和研究上极具有潜力,见图1.化合物Ⅰ及其盐是一类多巴胺受体,特别是对多巴胺D3受体具有良好的亲和力,由此能显著地调节多巴胺D3受体,此类化合物可能用作抗精神病药,例如用于治疗精神分裂症,精神病性抑郁症,躁狂症,偏执狂和妄想症等[1].化合物Ⅱ的细胞效能在治疗癌症的晚期临床试验中被发现有很强的体外抗增殖活性、良好的溶解性和可接受的CYP3A4外形(一类亚铁血红素),对该化合物的体外安全性进行评估,结果表明该化合物可用于进一步的先导优化[2].
内皮素是最初从血管内皮细胞中分离出来的有效的血管收缩和有丝分裂肽家族,已知这些肽通过与特定的G蛋白偶联受体相互作用,能引起许多心血管和肾脏功能障碍的生物学效应.化合物Ⅲ是异吲哚啉二羧酸类似物,研究发现这是一种高效的ETA型内皮素选择性受体拮抗剂[3].Ⅳ类化合物是一类淀粉样蛋白聚集的抑制剂,在一般情况下可以用于评估与淀粉样蛋白聚集有关的疾病,尤其是阿尔茨海默氏病,其在对病体的标准体内试验中显示出良好的活性[4].化合物Ⅴ是一种新型的Hsp90抑制剂,这种抑制剂可以抑制黑色素瘤细胞的增殖和锚定非依赖性生长,并消除体内异种移植瘤的生长;也可在黑素瘤细胞中诱导抗肿瘤活性,并且可能通过协同靶向多种途径在黑素瘤患者中显示出治疗益处[5].
异吲哚啉类化合物作为一种具有各种生物药理作用的生物碱,其多由人工合成得到,由于其独特的生理性质,已合成出许多新型异吲哚啉及其衍生物以进一步研究它们生理活性和构效关系,一系列异吲哚啉酮骨架单元在合成多种有用的有机分子作为关键的合成中间体而受到许多研究者的注目.
Geraldine Masson课题组[6]描述了一种新型的可见光光氧化还原促进的三组分串联方法,用于从易获得的起始原料合成含CF3的异吲哚啉;Shinobu Takizawa课题组[7]通过有机催化Betti/aza-Michael反应开发了一种高度经济,非对映性选择的方法合成了一系列重要的1,3-二取代异吲哚啉;姚和权课题组[8]开发了串联的1,6-共轭加成/1,4-Michael加成,用于在新设计的邻位亲电取代的对甲氧基醌类化合物和亲核试剂之间合成异吲哚啉类化合物,该方法具有温和的反应条件,高收率和良好的官能团耐受性;丁明武课题组[9]通过连续的Ugi/Aza-Michael加成反应,开发了一种具有两个立体中心的多取代异吲哚啉的新的高效非对映选择性合成方法;Christopher Hyland课题组[10]利用氨基苯甲醛与N-取代的α-氨基酸发生分子内(3+2)环加成热反应,通过脱芳香化/再芳香化顺序得到了各种取代的异吲哚啉类化合物;此外,还有众多高效新颖的合成方法对异吲哚啉类化合物进行研究[11-15].
本文研究了在无催化剂和无溶剂条件下合成异吲哚啉类化合物的合成方法,利用邻二氯苄和各类取代胺之间的熔融反应完成了这一系列的转化.本文的方法反应过程中不用任何的催化剂和溶剂,且后处理过程简便,有较强的底物适用性.
1 实验部分
1.1 仪器与试剂
核磁共振波谱仪,CDCl3-d1为溶剂,TMS为内标,Bruker-400 MHz spectrometer;高分辨质谱仪,Waters Xevo-G2 Q-Tof mass spectrometer;熔点仪,Beijing Tech X-4 micro melting point apparatus.
邻二氯苄,苯胺,正丁胺,苯甲胺,苯乙胺,2-氟苯胺,3-氟苯胺,4-氟苯胺,4-氯苯胺,4-溴苯胺,4-碘苯胺,4-异丙基苯胺,4-叔丁基苯胺,2-甲基苯胺,3-甲基苯胺,4-三氟甲基苯胺;均为分析纯,萨恩化学技术(上海)有限公司.
1.2 实验过程
异吲哚啉化合物3a-3o的合成,见图2,在50 mL的单口瓶中加入邻二氯苄(10 mmol,1.0 eq),各种取代胺2a-2o(10 mmol,1.0 eq)和三乙胺(20 mmol,2.0 eq),在80℃下搅拌1 h.期间用薄层色谱法(TLC)监测反应.反应完成后,将反应体系内的温度降到室温,加入30 mL的石油醚,搅拌10 min,过滤得到滤液,真空蒸发除去石油醚得到异吲哚啉类化合物3a-3o.
2 结果与讨论
尝试将邻二氯苄,苯胺和三乙胺在无溶剂无催化条件下进行反应,如图3所示.

图3合成异吲哚啉3aFig.3 Synthesis of isoindoline 3a
在室温下按摩尔量比1∶1∶2地加入邻二氯苄,苯胺和三乙胺,然后缓慢地梯度升温,期间用薄层色谱法(TLC)密切跟踪反应,监测到反应原料基本反应完全,并在薄层板上产生一个新的紫外点,此时温度为50℃,观察到相比于原料邻二氯苄,新的紫外点的极性很大,但又比原料苯胺的极性略小,推测是邻二氯苄与苯胺反应脱除了第一个HCl;保持反应温度不变,延长反应时间30 min,薄层板上又产生了一个新的紫外点,观察到新的紫外点的极性比原料邻二氯苄接近,推测是反应中间体分子内脱除了第二个HCl得到终产品;延长反应时间至1 h,直到新的紫外点足够多量,开始分离产物.将反应体系里的温度降到室温,加入一定量的石油醚,搅拌15 min,过滤得到滤液,真空蒸发除去石油醚,得白色固体,再用薄层色谱法进行初步判断,发现薄层板上只出现的一个单一点.然后表征该化合物的氢谱(1H NMR)和碳谱(13C NMR),并用高分辨质谱仪确定其准确的分子质量(HRMS),同时与已有报道的文献数据相对应分析,确定最终产物为2-苯基异吲哚啉.经过此反应的初步小试,确定无催化剂无溶剂条件熔融合成异吲哚啉类化合物有一定的可行性.
2.1 反应条件探索
以3a为反应模型底物,在无溶剂无催化的条件下,反应体系中的温度对反应过程中的反应速率有直接的影响,由于反应原料邻二氯苄的熔点比较低(51℃),苯胺在室温下也为液体,所以反应体系内的熔融状态为液态,混合均匀性也是良好,所以反应速率基本上只受到温度的影响.考虑到反应的顺利进行过程和高产率地得到终产物,将反应温度设定在50℃至90℃进行优化,实验结果如表1所示.反应温度80℃以下的反应速率相对较慢,即使在1小时后,反应体系中仍存在比较多的中间体,而80℃以上的产率在80%以上,反应中间体大部分能转化为产物.实验结果表明本合成方法能取得良好的收率,具有一定的原子经济性.由于反应结束后体系内的物质成分为产物,未转化完全的中间体以及三乙胺盐酸盐,且反应中间体和产物的极性差别很大,可以很容易地将产物分离出来并回收中间体.
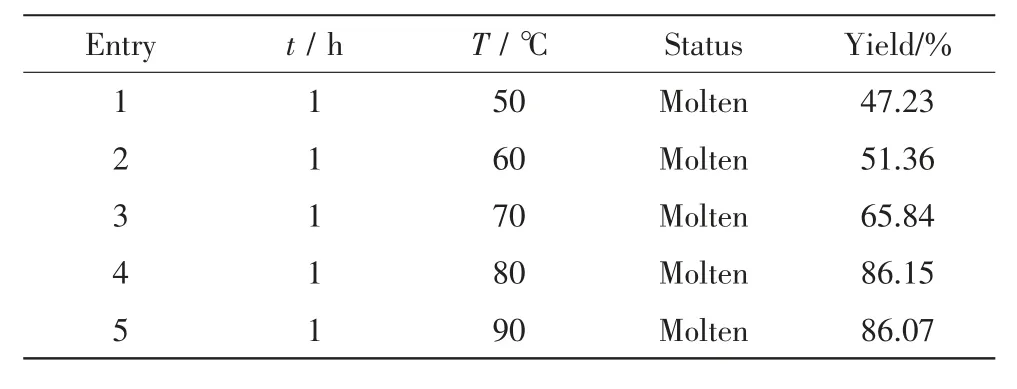
表1反应条件的优化Tab.1 Optimization of reaction conditions
2.2 底物适用性研究
在优化条件的温度下,选择了一系列取代胺和邻二氯苄进行反应,以验证该方法的适用性范围,实验结果如表2所示.
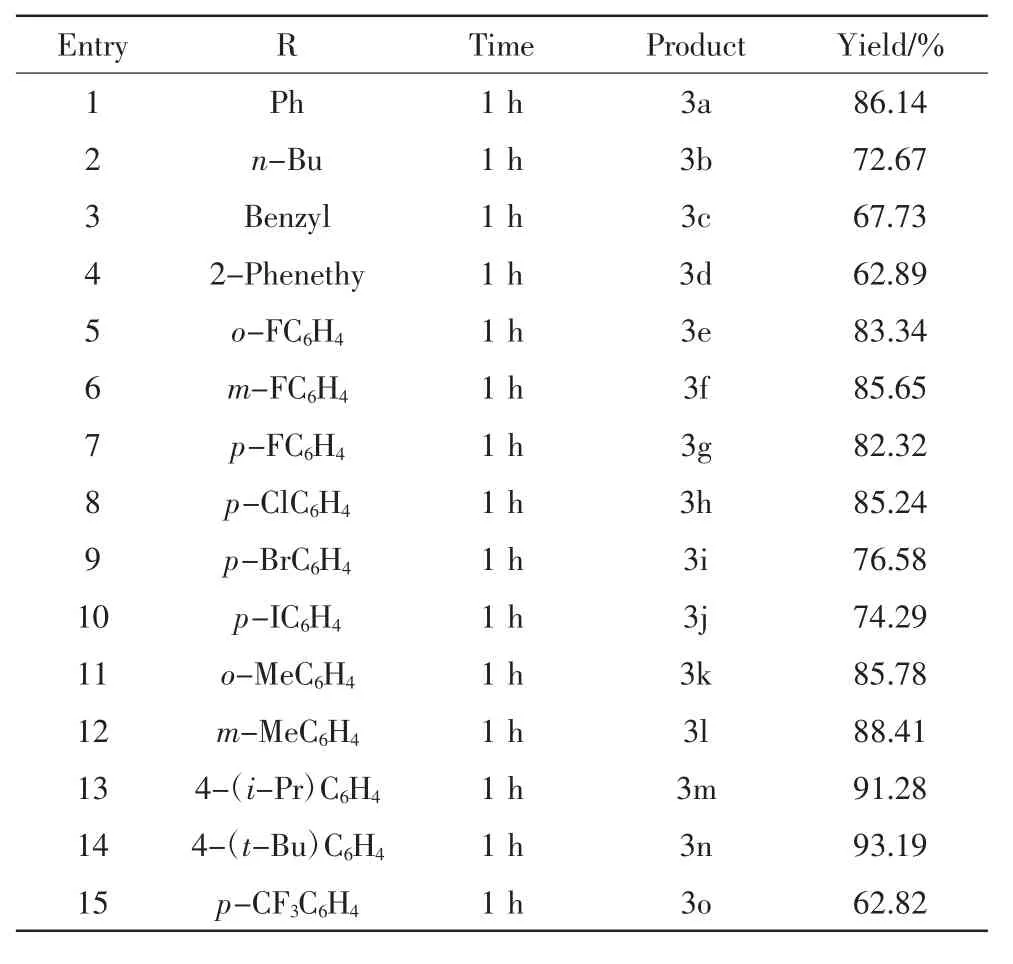
表2底物拓展Tab.2 Substrate expansion
在烷基类胺中(Entry2~4),设计了不同种类的胺,包括直链的正丁胺,苄胺和2-苯乙胺,这些基团有较好的反应活性,收率都超过了60%.实验结果还表明,带有卤素基团的芳香胺(Entry5~10)也能有效地进行反应,得到良好的收率,收率都超过了70%;另外在邻,间,对位置的氟基苯胺收率差别较小,表明具有不同取代位置的卤代苯胺不影响反应的进行.此外,在相同的反应条件下,苯环上带有烷基类的胺(Entry11~14),反应表现出了优良的效果,收率都超过了85%,最高达到了93%,推测是该类基团活化了苯环,促进了反应的进行.带有三氟甲基这一强吸电子的基团的苯胺(Entry15)也能有效地进行反应,收率也超过了60%,这也说明本方法对强吸电子的基团有一定的适用性.由于反应中间体与终产物的溶解性质差异较大(中间体难溶于石油醚,而终产物易溶于石油醚),反应产物可以轻易地从反应系统中分离出来.综上所述,本方法具有良好的底物适用范围和较好的官能团耐受性.
2.3 反应历程
以合成3a为例,如图4所示
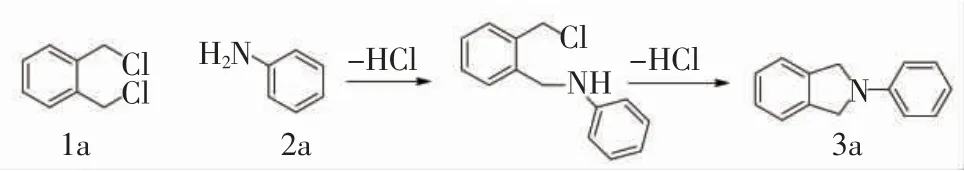
图4反应历程Fig.4 Reaction mechanism
邻二氯苄首先与苯胺反应脱除第一个HCl,形成仲胺中间体,随后该中间体再进行分子内关环脱除第二个HCl.通过对反应过程的监测发现,邻二氯苄与苯胺反应形成中间体基本是定量转化的,而在分子内关环步骤,由于对反应分子的活性要求较高,仲胺中间体并不能完全地脱除第二个HCl.不过由于终产物的极性远小于反应中间体的极性,所以能轻易地将二者分离开.
2.4 化合物表征
本文对目标化合物3a-3o进行了熔点测试,高分辨质谱,核磁氢谱以及碳谱的表征,实验数据如表3所示.同时在参照已有文献报道数据的基础上,确定为所要合成的目标化合物.

表3化合物表征Tab.3 Characterization of compounds
3 结论
本文研究了在无溶剂无催化条件下以邻二氯苄和各种取代胺合成一系列异吲哚啉类化合物的方法,该方法在反应过程中不使用任何的催化剂和溶剂,大大地降低了反应的用料成本,是对合成异吲哚啉类化合物方法学研究的重要拓展,具有较好的底物适用性,取得了不错的收率;且反应条件简单,只需加热反应即可,后处理过程简便,具有工业化前景.
猜你喜欢
能源化工(2021年6期)2021-12-30 15:41:26
云南化工(2021年10期)2021-12-21 07:33:24
中国资源综合利用(2017年4期)2018-01-22 02:46:55
当代化工研究(2016年7期)2016-03-20 16:21:54
合成化学(2015年9期)2016-01-17 08:57:20
船舶标准化工程师(2015年5期)2015-12-03 11:00:24
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
西部皮革(2015年15期)2015-02-28 18:14:36
中国氯碱(2014年10期)2014-02-28 01:04:59
现代检验医学杂志(2014年3期)2014-02-02 02:42:29
