
基于连接强度的动态蛋白质网络构建算法研究∗
2020-12-23 11:49罗爱静许家祺颜湘茹
计算机与数字工程 2020年11期
李 鹏 闵 慧 罗爱静 许家祺 颜湘茹 伊 娜 刘 杰
(1.湖南中医药大学信息科学与工程学院 长沙 410208)(2.中南大学湘雅三医院 长沙 410006)(3.医学信息研究湖南省普通高等学校重点实验室(中南大学) 长沙 410006)(4.湖南信息职业技术学院软件学院 长沙 410200)
1 引言
自从人类基因测序工程完成后,生命科学研究的重点已经从基因组学转到了蛋白组学[1]。同时随着计算机硬件的发展以及智能信息处理技术的进步,采用计算机相关技术对蛋白组学中的诸多问题展开分析和研究是目前的热点。其中,关于蛋白质相互作用网络(Protein-Protein Interaction Network,PPIN)[2~3]的研究是一项基础性的工作。
众所周知,生物体内蛋白质之间的相互作用总是动态变化的[4],这种变化体现着生命进化与发展的一种自然趋势和必然结果。然而,动态变化的蛋白质网络给基于计算机技术的蛋白组学研究带来巨大的挑战,如何准确地对动态蛋白质网络进行建模和分析已经成为制约该领域中很多问题研究的瓶颈。为此,国内外相关学者对蛋白质网络的建模问题进行了大量的研究,提出了一系列有代表性的建模方案,例如,文献[7]从多维角度出发综述了构建蛋白质网络的常见方法,并展望了动态蛋白质网络研究的发展趋势。文献[8]根据蛋白质的基因表达变化情况将蛋白质分为动态和静态两类,进而提出了一种动态-静态蛋白质混合的时序网络构建新方法。然而该方法缺少对噪音的系统化分析,网络构建结果容易受到假阳性和假阴性数据的干扰。文献[9]利用概率统计中常见的3-σ 法则来判断蛋白质的活性,进而提出了基于活性周期的蛋白质网络构建方法。但是这种方法经常会过滤掉一些一直有较高表达信息的蛋白质,造成数据的丢失。胡塞等[10]分析了蛋白质相互作用数据和基因表达数据对于网络构建的作用,建立了一种改进的动态蛋白质网络D-PIN(Dynamic Protein Interaction Networks)。然而该文对于采样周期的选择主要通过实验设定,不具有普适性。针对以上方法的不足,本文对动态蛋白质网络的构建问题进行了研究,提出了一种基于连接强度的动态蛋白质网络构建算法。并最后通过仿真实验验证了所提算法的有效性。
2 构建动态蛋白质网络
本文借鉴进化图[11]在描述复杂动态网络方面的优势,采用进化图来完成动态蛋白质网络建模过程。为了便于理解,下面给出一些相关的定义:
定义1 进化图假设有一动态图G=(V,E),V是G 的顶点,E 是G 的边。它的子图包含:GS={},有。设TS=t1,t2,…,tT表示所有子图存续时间,则称Θ=(G,GS,TSi)是进化图,其中i=1,2,…,T 。
定义2 活性蛋白质设Pr 表示某一生物体内的一个蛋白质,PrAGE表示Pr 的基因表达均值,如果在某一时间段T 内,都存在关系:PrAGE≥ε,其中ε 是阈值因子。则称Pr 是活性蛋白质,并记Ac(Pr)为Pr 的活性周期。
2.1 网络构建细节
紧接着上述定义,我们分为如下的三个阶段来构建动态蛋白质网络:1)基于基因表达均值计算来判断各个蛋白质的活性,确定各自的活性周期;2)对各个活性蛋白质划分时间片,具有相同活性周期的蛋白质拥有同一时间。对于同一时间的所有活性蛋白质,依据后续定义的连接强度来构建蛋白质子网;3)采用进化图理论对各个蛋白质子网进行建模,从而构建得到动态蛋白质网络。
2.1.1 计算蛋白质的活性周期
蛋白质活性周期的计算是构建动态蛋白质网络的第一步。假设,蛋白质Pr 在时刻i 的基因表达值为,1 ≤i ≤n。Pr 的基因表达值的标准差为(Pr)。则有如下的计算公式:

根据式(1)和式(2),文中定义了函数V(Pr)表示蛋白质Pr 的基因表达情况的变化:

一般而言,0 ≤V(Pr)≤1。紧接着,我们利用经典的3-sigma 准则[9]来确定活性阈值ε ,其计算公式为

对于任意给定的一个时间片,若有PrAGE(Pr1,Pr2,…,Prk)>ε(ε 为活性阈值),则认为这k 个蛋白质具有相同的活性,用它们来构建同一个蛋白质子网。对于生物体内的所有蛋白质而言,利用蛋白质活性计算可以统计得到具有不同活性周期的蛋白质集合S_Pr={T1,T2,…,Tk}。最后我们根据S_Pr 中元素的个数来决定划分出多少个蛋白质子网。
2.1.2 构建子网
计算得到所有蛋白质的不同活性之后,可以构建出不同的蛋白质子网。下面仅以其中的任意一个子网为例来阐述其构建过程。假设{Pr1,Pr2,…,Prl}表示具有相同活性的l 个蛋白质,现在对它们构建子网。要准确地构建出蛋白质子网的关键在于发现这l 个蛋白质的相互作用关系。文中通过定义连接强度这一个概念来对蛋白质之间是否具有相互作用来进行评价。具体而言,文中从两个方面考虑蛋白质与蛋白质之间的连接强度:1)公共邻居数量。如果两个蛋白质之间存在越多的公共邻居,这表明它们之间具有更为紧密的相互作用关系;2)边和度的比例。如果某两个蛋白质之间的邻接边越多,并且度越小。则它们之间具有更紧密的相互作用关系。综上所述,可以采用下面的公式计算连接强度:
定义3 连接强度
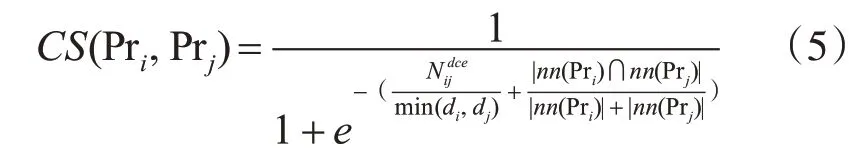
其中,CS(Pri,Prj)表示任意两个蛋白质Pri和Prj之间的连接强度;表示Pri和Prj之间存在的邻接边个数;nn(Pri)表示Pri的邻居节点;di表示Pri的度;式(5)中的是一个Sigmoid 函数[12],使用该函数的好处在于:它可以将影响蛋白质之间相互作用强弱的诸多因素(邻接边个数、节点的度等)最终转为一个概率值,能够较好地刻画不同蛋白质之间的连接关系。
2.2 动态蛋白质网络构建算法描述
相对于静态蛋白质网络而言,动态蛋白质网络的拓扑结构会随着蛋白质合成或降解、生物环境等因素的变化而动态变化。对蛋白质网络准确建模的关键是采用合适的模型来表示这个动态变化因素。考虑到网络中大多数蛋白质的基因表达具有时间周期特性,并不是完全随机的,因此文中从时间维度出发对动态蛋白质网络进行建模,首先基于时间片的概念对整个网络进行划分,定义出每个时间片内的网络连通性,然后基于进化图理论将多个时间片内的子网构建成动态蛋白质网络模型,具体细节见算法1。
算法1 动态蛋白质网络构建算法(DPPN-CC)
输入:基本表达值数据,PPI数据,阈值th
输出:动态蛋白质网络模型Θ=(G,GS,TSi)
步骤1. 根据所有蛋白质的基因表达值数据,采用式(1~3)计算生物体内所有蛋白质的活性周期Ac(Pr),并采用列表对其结果进行存储,可得:
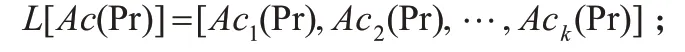
步骤2.根据蛋白质的活性周期来构造子网:
For Aci(Pr),i=1,2,…,k in L[Ac(Pr)]:
在Aci(Pr)中计算CS(Pri,Prj);
If CS(Pri,Prj)≥th,则在Pri和Prj之间增加边<Pri,Prj>,并记录<Pri,Prj>所在的时间片TSi;
步骤3.如果L[Ac(Pr)]不为空,则重复执行步骤2;否则算法终止。
3 实验结果与分析
下面以蛋白质复合物的识别作为测试应用,在经 典 的DIP 数 据 集[13]和CYC2008 数 据 集[14]上 对DPNC-CC 算法的性能进行了评价。其中,算法的实现采用Python语言;评价指标采用:查全率、查准率和F-measure。仿真实验环境为:64 位的Windows10操作系统+anaconda平台。
3.1 参数敏感性分析
从算法1中的描述可知,参数th 的取值大小直接影响着构建出来的动态蛋白质网络的拓扑结构,因此为了衡量DPNC-CC 算法的可靠性,有必要对该算法的参数敏感性做出详细的分析。我们以CYC2008数据集为测试数据集,在构建出来的网络上依次运行MPC-TPW[15]和DPC-NADPIN[16]等两种复合物识别算法,采用F-measure 指标来评价DPNC-CC 算法的性能。实验结果见图1。仔细观察图1 可以发现,随着th 取值的增大,两种识别算法的识别性能也在逐步上升,但当th 取值超过0.7之后,两种识别算法的F-measure 值基本不再波动,这表明通过DPNC-CC 算法构建的动态蛋白质网络不具有参数敏感性,可以推广到蛋白组学的众多应用问题中去。
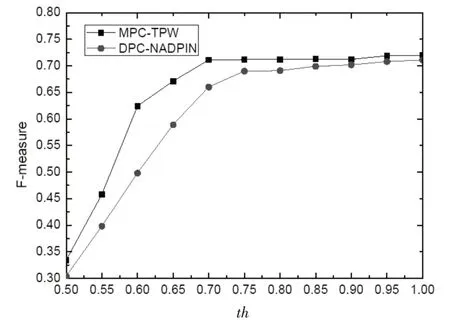
图1 DPNC-CC算法的参数敏感性分析
3.2 DPNC-CC算法与其他算法的比较
以DIP 数据集为实验对象,下面以DPNC-CC算法与文献[4~6]中的算法构建得到的动态蛋白质网络上运行MPC-TPW 算法进行复合物识别,来测试不同的网络构建算法的有效性。文中采用K 折交叉验证(K=10)来进行仿真实验,取10 次实验结果的均值作为各个算法在DIP 数据集的复合物识别结果,见表1。

表1 MPC-TPW算法在各个网络上的性能比较
从表1 可以看到,MPC-TPW 算法在本文构建的动态蛋白质网络(DPNC-CC)上进行复合物识别的查全率和查准率都要优于另外的四种算法。F-measure 值要比文献[4]的算法、文献[5]的算法和文献[6]的算法分别高约53%、24%和21%。这主要是因为:本文算法在构建动态蛋白质网络时,不仅从物理上考虑了蛋白质与蛋白质之间的距离、拓扑结构等信息对网络构建的影响,还利用了蛋白质的活性周期这一生物信息来衡量蛋白质之间的相互作用关系,较为全面地规避了蛋白质网络中可能存在的虚假信息,从而能够更好地识别蛋白质复合物。这也从侧面印证了DPNC-CC算法构建的动态蛋白质网络更优。
3.3 鲁棒性分析
下面进一步对DPNC-CC 算法在包含噪声(假阳性和假阴性)的蛋白质相互数据集上的性能表现进行实验分析。首先,我们通过在已经构建好的蛋白质网络上随机增加一定比例的边数来模拟数据的假阳性。边数每次增加20%,增加的尺度从20%上升到100%,可以得到五组包含假阳性的蛋白质相互作用数据,然后采用DPNC-CC 算法对这五组数据进行复合物的识别,识别结果的查准率和查全率如图2 所示。从图2 可以明显观察到,数据假阳性的增加,只会轻微降低DPNC-CC算法的查准率,对于DPNC-CC算法的查全率基本没有影响。
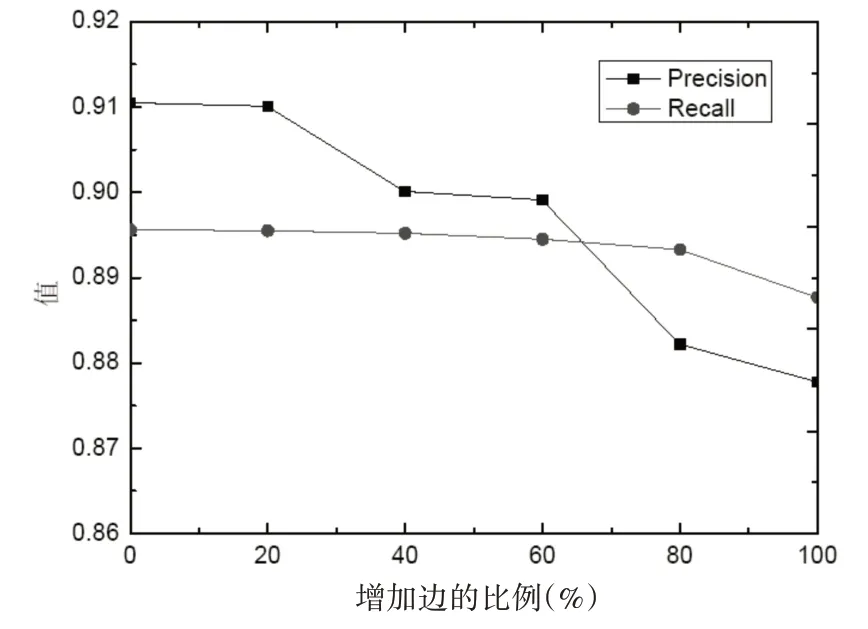
图2 数据包含假阳性时的DPNC-CC算法性能
最后,我们再次在已经构建好的蛋白质网络上随机删除一定比例的边数来模拟数据的假阴性。删除的边数每次增加20%,增加的尺度从20%上升到100%,可以得到五组包含假阴性的蛋白质相互作用数据,然后采用DPNC-CC 算法对这五组数据进行复合物的识别,识别结果的查准率和查全率如图3 所示。从图3 可以明显观察到,随着数据假阴性的增加,DPNC-CC 算法在前期的查全率和查准率基本保持不变,但当删除的边的比例超过45%之后,DPNC-CC 算法的识别质量则呈现着明显下降的趋势,这主要是由于随着边的删除将会使得蛋白质相互作用数据中大量真实存在的相互作用被删除,从而导致算法的识别结果大大地降低。总的来看,本文算法在包含噪声的蛋白质相互作用数据集中的表现是可信的,算法能够对数据的动态变化做出正确响应,具有较好的鲁棒性。
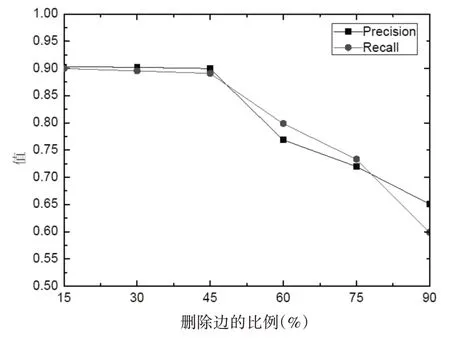
图3 数据包含假阴性时的DPNC-CC算法性能
4 结语
蛋白质网络的构建是蛋白组学中众多问题研究的基础,文中针对现有构建算法存在的不足,提出了一种基于连接强度的动态蛋白质网络构建算法,并通过仿真实验验证了该方法在蛋白质复合物识别上的有效性。下一步,我们将在本文的基础上进一步对动态蛋白质网络中的复合物挖掘问题展开研究,力争为生物学家或医学家的工作提供更多的技术支撑。
猜你喜欢
卫星应用(2022年7期)2022-09-05
九江学院学报(自然科学版)(2022年2期)2022-07-02
中国海洋大学学报(自然科学版)(2022年7期)2022-06-28
卫星应用(2022年3期)2022-05-23
卫星应用(2022年1期)2022-03-09
中草药(2022年5期)2022-03-03
中国生物化学与分子生物学报(2022年1期)2022-02-26
环球慈善(2019年6期)2019-09-25
大学教育(2019年5期)2019-04-23
中国新通信(2019年21期)2019-03-30
