
红曲菌MpigE基因过表达质粒的构建
2020-12-21 03:51郑学海柳燕朱锦懋杨成龙陈由强陈建楠
福建农业科技 2020年5期
郑学海 柳燕 朱锦懋 杨成龙 陈由强 陈建楠

摘 要:以福建红曲菌MCL为出发菌株,以PSKH质粒为模板克隆PtrpC启动子及TrpC终止子。通过重叠延伸PCR技术融合PtrpC启动子、MpigE(目的)基因、TrpC终止子,获得PMT融合片段。为了验证融合片段PMT的正确性,进行琼脂糖凝胶电泳分析,在2100 bp左右出现单一且清晰的条带。通过EcoR V和Spe I酶切PMT片段,插入带有潮霉素抗性的PSKH质粒的EcoR V和Spe I位点,构建1个红曲菌过表达载体HPMT,大小为7 155 bp。通过构建过表达载体,以期对红曲菌MCL进行改造,获得高产色素低产桔霉素的菌株。
关键词:红曲菌;MpigE基因;质粒;过表达;构建
中图分类号:TS261.3 文献标志码:A 文章编号:0253-2301(2020)05-0001-11
DOI: 10.13651/j.cnki.fjnykj.2020.05.001
Abstract: By taking the MCL of Monascus in Fujian as the original strain, the PtrpC promoter and TrpC terminator were cloned by using the PSKH plasmid as the template. The PMT fusion fragments were obtained by fusing PtrpC promoter, MpigE target gene and TrpC terminator with the overlap-extension PCR technology. In order to verify the correctness of the fusion fragment PMT, the agarose gel electrophoresis analysis was carried out, and a single clear band appeared at about 2,100 bp. The fragment of PMT was digested by EcoR V and Spe I, and the EcoR V and Spe I sites of the PSKH plasmid with hydthromycin resistance were inserted to construct an overexpression vector HPMT of Monascus, with a size of 7 155 bp. By constructing the overexpression vector, the MCL of Monascus was modified, thus to obtain the strain with high yield of pigment and low yield of citrinin.
Key words: Monascus; MpigE gene; Plasmid; Overexpression; Construct
紅曲霉是目前世界上唯一生产食用色素的微生物,广泛应用于制醋、制酒、酱油、饮料、腐乳、香肠、肉制品、蜜饯等食品中[1-4]。但自从1995年法国Blance等[5]在红曲霉的发酵过程中发现了桔霉素后,红曲产品的安全性问题引起人们的广泛关注。利用传统的诱变、控制发酵条件等无法从根本上去除红曲发酵过程中的桔霉素,因此,人们逐渐将目光转向构建基因工程菌这一途径[6]。
目前基因的过表达技术在米曲霉、酵母菌等其他真菌中有较多的应用,但在红曲菌中的应用还相对较少。2005年秦丽娜[7]成功构建了酿酒酵母整合表达载体pUPGKAT(包括PGK强启动子、酿酒酵母乙醇脱氢酶基因Ⅰ和CYC1终止子),并将其转入原始菌株YS2△adh2重组菌株,结果表明构建的新菌株乙醇产量较出发菌株提高了8.84%。2009年王斌等[8]构建了米曲霉的重组表达载体pNMA-RML,并转入米曲霉宿主菌A.oryzaeniaD300,得到整合型的阳性转化子A.oryzaeONL。由于红曲的次级代谢产物红曲色素、桔霉素在合成途径上有相关性,具有共同的前体,如果对产红曲色素的相关基因进行过表达可能可以增强红曲产色素支路的代谢力度而降低产桔霉素的代谢力度,从而获得高产色素而低产桔霉素的菌株,达到利用基因工程进行分子育种的目的。目前已有研究表明pigR基因、MpigE基因均与红曲色素的合成具有相关性[9-10]。
基因超表达技术作为一项重要的分子生物学技术,是将目的基因的全长序列与高活性的组成型强启动子或组织特异性强启动子融合,从而构建功能型表达载体,通过转化获得该基因产物大量积累的菌株[11-14]。在构建超表达载体时,为了增强外源基因表达及增加表达载体在宿主内的稳定性需要慎重选择启动子及质粒。本研究所选用的启动子为PtrpC启动子,通过重叠延伸PCR技术融合PtrpC启动子、MpigE(目的)基因、TrpC终止子,并将融合的片段插入带有潮霉素抗性的PSKH质粒,构建一个红曲菌过表达载体。后期将再通过PEG介导原生质体转化构建出基因工程菌,获取高产色素低产桔霉素的菌株,从根本上解决红曲产品的桔霉素含量超标问题。
1 材料与方法
1.1 主要试剂材料
1.1.1 菌种与质粒 红曲霉MCL由实验室合作公司馈赠、DH5α大肠杆菌菌株由本实验室保存、PSKH质粒(带有PtrpC启动子、TrpC终止子及Hygromycin B抗性基因)由华中农大馈赠、pMD 19T 载体购自Takara(大连)有限公司。
1.1.2 试验试剂 Ex Taq酶、T4 DNA连接酶、EcoR V限制性内切酶、Spe I限制性内切酶、DNA Marker购自Takara(大连)有限公司;RNaseA、潮霉素购自Sigma公司;Fungal DNA Kit 购自Omega公司;胶回收试剂盒购自Promega公司;氨苄青霉素购自生工生物工程(上海)股份有限公司产品;PCR扩增引物均由Primer Premier 5軟件设计完成后由生工生物工程(上海)股份有限公司合成。
1.1.3 主要培养基 麦芽汁(MES)液体培养基:准确称取麦芽膏粉13.1 g溶于100 mL蒸馏水,pH自然,121℃灭菌15 min。麦芽汁(MES)固体培养基:准确称取麦芽膏粉13.1 g,琼脂2 g,溶于100 mL蒸馏水,pH自然,121℃灭菌15 min。MPPY培养基:4%葡萄糖,0.3%NaNO3,0.2%酵母浸粉,0.05%KCl,pH 6.8,121℃灭菌15 min。LB液体培养基:肉汤培养基2.5 g溶于100 mL蒸馏水,pH自然,1×105 Pa、121℃、灭菌20 min。LB固体培养基:琼脂培养基3.4 g溶于100 mL蒸馏水,pH自然,1×105 Pa、121℃、灭菌20 min。
1.1.4 主要仪器设备 微波炉(GRANT);洁净工作台(苏州市安泰空气技术有限公司);数显三用恒温水浴箱(上海比朗仪器有限公司);全自动新型鼓风干燥箱
ZFD5090(上海智城分析仪器有限公司);紫外可见分光光度计(上海美谱达);PCR仪(美国 Applied Biosystems);梯度PCR仪(美国 Applied Biosystems);连接仪(DRY BATH);凝胶成像系统(上海培清有限公司)。
1.2 试验方法
1.2.1 红曲菌孢子悬液的制备 (1)将实验室斜面保存的红曲菌MCL转接至MES固体培养基中,30℃划线培养3~4 d,待长出白色的单菌落。(2)挑取单菌落接种于MES液体培养基中,30℃、160 r·min-1,振荡培养2 d。(3)吸取200 μL菌液涂布MES固体培养基,28℃静置培养7~10 d。(4)待长出大量孢子后,用灭过菌的超纯水轻轻洗脱孢子,并用500目双层尼龙布过滤菌丝体。(5)用血球记数板测定孢子悬液浓度。
1.2.2 红曲菌基因组DNA的提取 红曲菌总DNA的提取采用Fungal DNA Kit进行提取,具体步骤如下:(1)按照2.3.2 培养红曲菌菌丝,用500目尼龙布过滤收集菌丝体。(2)灭菌的无菌水洗涤3次,滤纸挤压干水分,将菌丝放入预冷的研钵中,加入适量液氮迅速研磨成粉末状。(3)将粉末分装至2 mL离心管中(约1/3),立即加入600 μL Buffer FG1,震荡摇匀。65℃水浴10 min(水域期间震荡2~3次)。(4)加入140 μL Buffer FG2,震荡摇匀,12000 r·min-1、4℃、离心10 min。(5)小心吸取上清液至新的离心管中,加入0.7体积的异丙醇,震荡摇匀(这个步骤将去除大部分的多糖分子,通过提高DNA黏合物的容量来增加吸附柱的能力),12000 r·min-1、4℃、离心2 min。弃上清液,倒置离心管于滤纸上,1 min。(6)加入65℃遇热的300 μL无菌去离子水,震荡摇匀,65℃水浴短时间,促进DNA 溶解,加入4 μL RNase,加入150 μL Buffer FG3,加300 μL无水乙醇,震荡摇匀。(7)将所有试样移至有柱子的离心管中(试剂盒自带),12000 r·min-1、4℃、离心1 min。(8)将吸附柱移至新的离心管中,加入稀释过的DNA Wash Buffer 700 μL,12000 r·min-1、4℃、离心1 min,弃收集管中的液体。(9)重复步骤11。(10)以最高转速进行空离2 min。(11)将柱子移至1.5 mL的离心管中,加入65℃预热的无菌去离子水50 μL,室温放置2~3 min,12000 r·min-1、4℃、离心2 min。
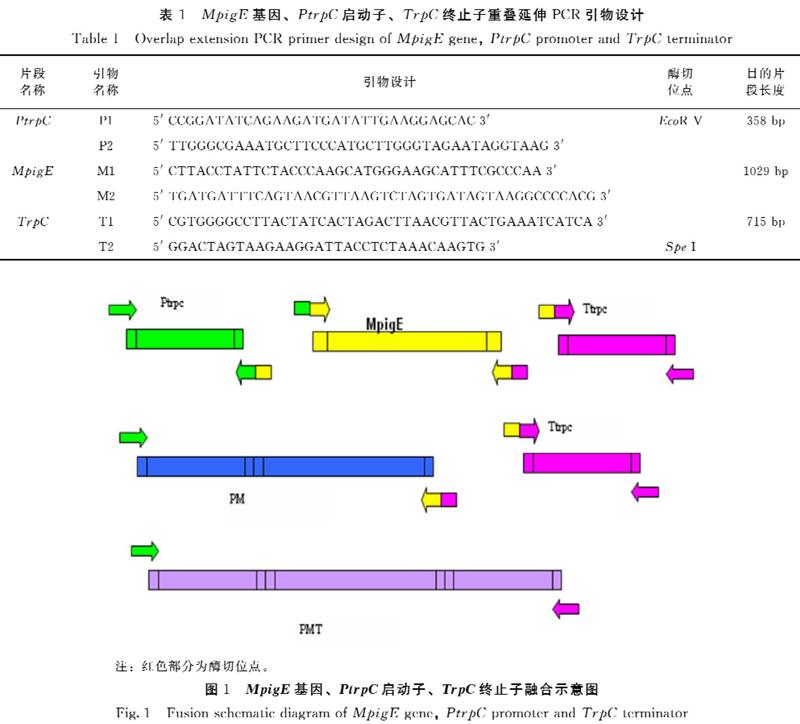
1.2.3 重叠延伸PCR引物设计 通过查找GenBank中公布的红曲菌MpigE基因序列、PtrpC启动子序列、TrpC终止子序列,利用重叠延伸PCR原理设计3对叠套引物用于PtrpC、MpigE、TrpC这3个片段的扩增与融合。引物序列、酶切位点、产物长度见表1、图1。引物由生工生物工程(上海)有限公司合成。
1.2.4 MpigE基因片段、PtrpC启动子片段、TrpC终止子片段的扩增 重叠延伸PCR需要进行两轮PCR,第一轮通过普通PCR程序进行扩增,依次获得需要融合的目的片段。以红曲菌基因组DNA为模板,用引物M1、M2扩增MpigE基因片段;以PSKH质粒为模板,用引物P1、P2扩增PtrpC启动子片段,引物T1、T2扩增TrpC终止子片段。
1.2.5 PtrpC启动子片段与MpigE基因片段的融合和PM片段与TrpC片段的融合 以PtrpC启动子片段与MpigE基因片段为模板,P1为上游引物,M2为下游引物,采用重叠延伸PCR的方法进行两片段的融合PM片段;再将PM片段与TrpC片段的融合片段称之为PMT片段。PMT片段再与pMD19T载体连接构成重组质粒PMT19,转化DH5α感受态细胞,送往上海生工进行测序分析。
1.2.6 红曲菌过表达载体的构建 红曲菌过表达载体构建见图2。
1.2.6.1 PMT19质粒的双酶切 将PMT19质粒用EcoR V和Spe I两种限制性内切酶按照表2酶切体系进行双酶切。
反应温度:37℃;反应时间:4~5 h。酶切产物用1%琼脂糖凝胶电泳验证,验证成功后扩大酶切体系为50 μL,并用切胶回收试剂盒目的片段。
1.2.6.2 PSKH质粒的EcoR V、Spe I双酶切 因为在PSKH质粒上EcoR V和Spe I两种限制性内切酶的酶切位点相距较近,因此采用分步酶切的方法。
(1)用限制性内切酶EcoR V按照表3酶切体系进行酶切。
反应温度:37℃;反应时间:4~5 h。
(2)用限制性内切酶Spe I按照表4的酶切体系进行酶切。
反应温度:37℃;反应时间:4~5 h:酶切产物用1%琼脂糖凝胶电泳验证,验证成功后扩大酶切体系为50 μL,并用胶回收试剂盒方法回收双酶切后的质粒。
(3)重组过表达载体HPMT。经过双酶切的PMT片段与用相同内切酶酶切的PSKH质粒连接成为过表达载体HPMT,其连接体系如表5。
(4)红曲菌过表达载体HPMT的鉴定。①重组质粒电泳大小鉴定。质粒提取用1%琼脂糖凝胶电泳验证,检测条带的位置,并以质粒PSKH作对照,比较两个质粒大小差异,判断PMT片段是否插入。②重组质粒酶切鉴定。按表2的双酶切体系进行验证。
2 结果与分析
2.1 红曲菌MCL培养效果及其总DNA提取
从红曲菌MCL的培养效果(图3)可以看出红曲菌MCL在MES培养基上长势良好,菌落蔓延幅度较大,菌落结构有不规则的辐射纹。从其长势可断定,已经产生孢子。按照Fungal DNA Kit所示步骤提取红曲菌基因组DNA,并进行凝胶电泳检测见图4。
2.2 MpigE基因片段、PtrpC启动子片段、TrpC终止子片段扩增鉴定结果
以红曲菌MCL基因组DNA为模板,以M1和M2为上下游引物,分别在50.0℃、50.8℃、51.9℃、53.3℃、54.8℃、56.5℃、58.1℃、59.7℃、61.0℃、61.9℃不同退火温度下扩增MpigE基因片段,结果见图5。
从图5可以看出在10个不同的退火温度下均在1000 bp偏上处扩增出一条清晰、单一的目的条带,扩增产物大小与目的片段基本完全一致。将扩增到的MpigE基因片段送往上海生工测序,测序结果见图6,输入NCBI进行blast分析,其测序结果与Genebank中公布的序列100%同源。
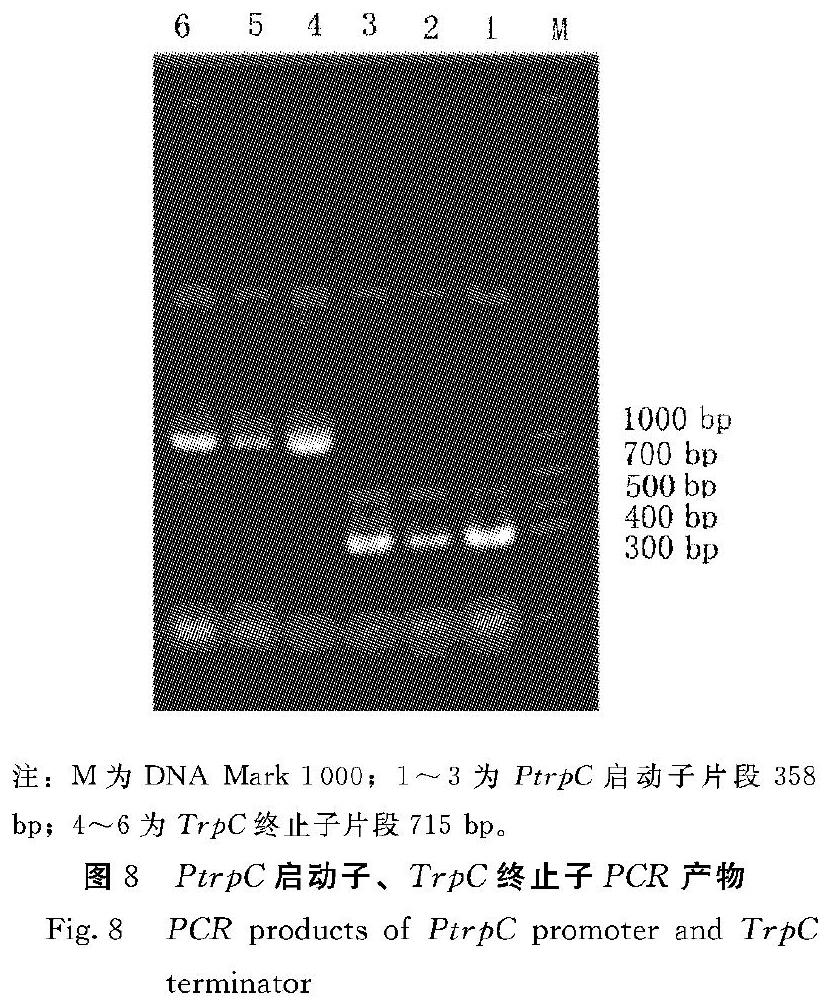
提取PSKH质粒,其电泳图谱见图7。以提取的PSKH质粒为模板, P1和P2為上下游引物扩增PtrpC启动子片段,T1和T2为上下游引物扩增TrpC终止子片段。退火温度设置为:55℃、58℃、60℃,其结果见图8。
从图7可以看出,PSKH质粒的提取成功,约在5000 bp处看到清晰的条带,与PSKH质粒的5081 bp一致。从图8可以看出,在3个不同的退火温度下均分别在约400 bp和700 bp处扩增出目的条带,扩增产物大小与PtrpC启动子片段、TrpC终止子片段大小基本一致,但退火温度为55℃及60℃时其条带更为清晰。将扩增到的PtrpC启动子片段、TrpC终止子片段送往上海生工测序,测序结果见图9、10,输入NCBI进行blast分析,其测序结果与Genebank中公布的序列100%同源。
2.3 PtrpC启动子片段与MpigE基因片段融合的鉴定结果
以PtrpC启动子片段与MpigE基因片段为模板,P1为上游引物,M2为下游引物,采用重叠延伸PCR的方法进行两片段的融合,退火温度设置为:50℃、55℃、58℃、60℃,其结果见图11。从图11可以看出,在以50℃、58℃的退火温度下均在约1500 bp处扩增出清晰、单一的目的条带,扩增产物大小与预期完全吻合。
2.4 PM片段与TrpC片段融合的鉴定结果
以PM片段与TrpC片段为模板, P1和T2为上下游引物,分别在50.0℃、51.9℃、53.3℃、54.8℃、56.5℃、58.1℃、59.7℃、61.0℃、61.9℃这9个不同的退火温度下扩增PMT片段,结果见图12。从图12可以看出9个不同的退火温度下均在约2100 bp处扩增出清晰、单一的目的条带条带,扩增产物大小与预期完全吻合,以50℃为退火温度时其目的条带最亮。
2.5 融合片段PMT的鉴定结果
将融合得到的PMT片段连入pMD19T 载体中构成重组载体PMT19,其电泳结果如图13所示。用EcoR V和Spe I两种限制性内切酶对载体PMT19进行双酶切,其电泳结果如图14所示。从图13和图14可以看出在5000 bp偏下处有清晰的条带,这与PMT19的大小一致。pMD19T 载体的大小为2692 bp,EMP片段的长度为2102 bp,从图15可见2500 bp上下各有1条清晰的条带,因此可以初步表明PMT片段克隆成功。
2.6 过表达载体PSKH构建结果
2.6.1 过表达质粒HPMT菌液PCR鉴定结果 用EcoR V和Spe I两种限制性内切酶对载体PMT19进行双酶切获得具有黏性末端的PMT片段,用相同的限制性内切酶酶切PSKH质粒,用T4连接酶连接构成重组质粒HPMT,转入大肠杆菌DH5α,以M1、M2为上下游引物进行菌液PCR验证。其电泳结果如图16所示。初步验证PMT片段与PSKH质粒连接成功。
2.6.2 过表达质粒HPMT电泳鉴定结果 为了进一步验证重组载体HPMT的正确性,用EcoR V和Spe I两种限制性内切酶进行双酶切验证。双酶切后应出现1条2102 bp大小的片段和1条5018 bp大小的片段。双酶结果如图17所示,从图17可以看出目的条带的大小都与预期相符,由此可知过表达载体构建成功。
3 结论与讨论
本试验以福建红曲菌MCL为出发菌株,成功提取该红曲菌基因组DNA,以此为模板克隆过表达的目的片段。以PSKH质粒为模板克隆PtrpC启动子及TrpC终止子。通过重叠延伸PCR技术融合PtrpC启动子、目的基因、TrpC终止子,该方法避免了采用限制性内切酶消化和连接酶连接等繁复的过程,直接通过多次PCR反应便可得到目的片段。为了验证融合片段PMT的正确性,进行琼脂糖凝胶电泳分析,在2100 bp左右出现单一且清晰的条带,初步验证融合成功。进一步将PMT片段与pMD19T载体连接转化大肠杆菌Dh5α,送往生工生物工程(上海)股份有限公司进行测序分析,测序结果表明所克隆的融合片段PMT完全正确。通过EcoR V和Spe I酶切PMT片段,插入带有潮霉素抗性的PSKH质粒的EcoR V和Spe I位点,构建1个红曲菌过表达载体HPMT,大小为7155 bp,并通过琼脂糖凝胶电泳鉴定及质粒双酶切验证表明过表达载体构建成功。后期将再通过PEG介导原生质体转化构建出基因工程菌,获取高产色素低产桔霉素的菌株,从根本上解决红曲产品的桔霉素含量超标问题。
参考文献:
[1]沈士秀.红曲的研究、生产及应用[J].食品工业科技,2001,22(1):85.
[2]李金红.红曲的生产及其功能和应用[J].中国调味品,2006(5):40-42.
[3]PETRA PATAKOVA.Monascus secondary metabolites:production and biological activity[J].J Ind Microbiol Biotechnol,2013(40):169-181.
[4]YAN L F,YAN CH SH,FU SH CH.Monascus pigments[J].Appl Microbiol Biotechnol,2012,96:1421-1440.
[5]BLANC P J,LAUSSAC J P,BARS J LE,et al.Characterization of monascidin A from Monascus as citrinin[J].International Journal of Food Microbiology,1995,27(2-3):201.
[6]王文鳳,袁兵兵,徐玲.红曲的研究现状及发展趋势[J].中国饲料添加剂,2014(2):1-7.
[7]秦丽娜.酿酒酵母乙醇脱氢酶Ⅰ基因的超表达[D].福州:福建师范大学,2005.
[8]王斌,潘力,郭勇.丝状真菌米曲霉外源基因表达系统的构建[J].华南理工大学学报,2009,37(6):84-89.
[9]赖卫华,许杨,熊勇华.红曲菌cDNA消减文库的构建[J].菌物系统,2003,22(3):466-473.
[10]SHIMIZU T,KINOSHITA H,ISHIHARA S,et al.Polyketide synthase gene responsible for citrinin biosynthesisin Monascus purpureu[J].Appliedand Environmental Microbiolog,2005,71(7):3453-3457.
[11]付桂明.橙色红曲菌pksCT gene的敲除和长同源臂置换型打靶载体的构建[D].南昌:南昌大学,2007.
[12]SHIMIZU T,KINOSHITA H,NIHIRA T.Identification and in vivo functional analysis by gene disruption of ctnA,an activator gene involved in citrinin biosynthesis in Monascus purpureus[J].Applied and Environmental Microbiology,2007,73(16):5097-5103.
[13]XIE N N,LIU Q P,CHEN F S.Deletion of pigR gene in Monascus ruber leads to loss of pigment production[J].Biotechnol Lett,2013,(35):1425-1432.
[14]LIU Q P,XIE N N,HE Y,et al.MpigE,a gene involved in pigment biosynthesis in Monascus ruber M7[J].Biotechnol Lett,2014,98:285-296.
(责任编辑:柯文辉)
猜你喜欢
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
中学生物学(2017年11期)2018-01-16
科学与财富(2016年28期)2016-10-14
大学教育(2016年9期)2016-10-09
成才之路(2016年26期)2016-10-08
成才之路(2016年25期)2016-10-08
安徽理工大学学报·自然科学版(2015年2期)2015-08-19
安徽理工大学学报·自然科学版(2015年1期)2015-07-21
安徽理工大学学报·自然科学版(2015年1期)2015-07-21
