
组蛋白赖氨酸甲基转移酶NSD3在肿瘤发生中的作用*
2020-12-17 08:25张瑾李艳潘云
中国肿瘤临床 2020年22期
张瑾 李艳 潘云
NSD3[nuclear receptor suppressor of variegation,enhancer of zeste,and trithorax(SET)domain-contain⁃ing 3]是组蛋白赖氨酸甲基转移酶(histone lysine methyltransferase,HKMTases)NSD(nuclear receptorbinding SET Domain)家族成员中的一员,该家族由NSD1、NSD2(MMSET/WHSC1)和NSD3(又称Wolf-Hirschhorn syndrome candidate 1-like 1,WHSC1L1)组成。NSD3 可识别并甲基化组蛋白赖氨酸残基,以调节染色质的完整性和基因的表达,越来越多的研究证实了NSD3 基因的扩增或与其他基因的融合等与人类肿瘤的发生密切相关,本文对NSD3在多种肿瘤中的研究作一综述,综合阐述NSD3结构特征及其在肿瘤发生中的作用机制。
1 NSD3蛋白的结构与特征
NSD3于2001年因蛋白质C端有700个氨基酸与NSD1、NSD2 相同,而被发现。其基因位于8p11.23上,迄今为止,已证实了NSD3 通过不同的剪切方式可衍生出3 种异构蛋白:长(long,L)、短(short,S)和WHISTLE蛋白(图1)[1-2]。除了NSD3S外均具有SET结构域、PWWP 结构域(proline-tryptophan-trypto⁃phan-proline,脯氨酸-色氨酸-色氨酸-脯氨酸)、PHD锌指结构域(plant homeodomain)。NSD3L 特有的C端富含半胱氨酸-组氨酸(cys-his-rich domain,C5HCH)结构[3-5]。NSD3L、NSD3S 在大脑、心脏和骨骼肌中高表达[2],是人类重要的致癌基因,过表达能将健康细胞转化为癌细胞[6]。WHISTLE 主要在人睾丸和急性淋巴细胞白血病中表达,有报道显示其可以抑制基因转录[7]。
SET结构域可细分为前SET结构域,核心SET结构域、后置SET 结构域,其利用辅因子S-腺苷-L-甲硫氨酸介导NSD 家族蛋白的甲基转移酶活性,是NSD3 发挥催化作用的功能基团。NSD3 C 末端(Cterminal domain,CTD)的SET结构域可以在核小体上甲基化组蛋白H3 赖氨酸K36 残基;后置SET 结构域中的TKKKTR 序列是与核小体的结合位点,以核小体为底物催化组蛋白H3 赖氨酸K36 二甲基化(his⁃tone H3 lysine K36 dimethylation,H3K36me2)[8-9]。
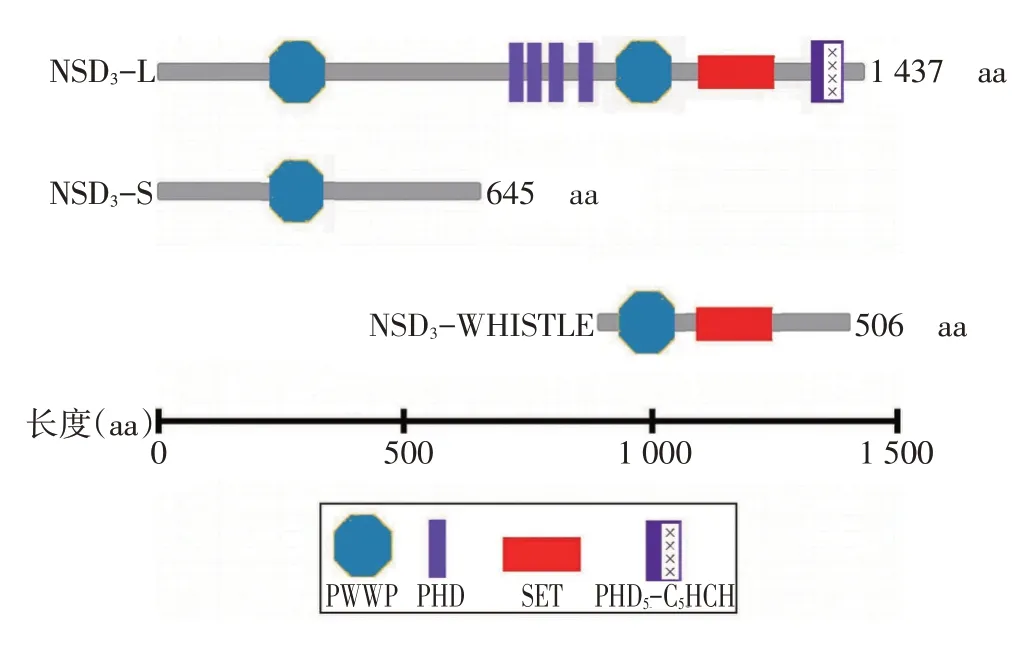
图1 组蛋白赖氨酸甲基转移酶NSD3结构图
PWWP结构域通过在核小体水平识别DNA和组蛋白赖氨酸的甲基化来充当染色质修饰的解读器[6,10]。PWWP结构域具有双重功能,既能与DNA结合,又能与甲基化的组蛋白赖氨酸结合。通过实验发现该结构域直接与H3K36 三甲基化(trimethyl⁃ation,me3)结合,而与H3K36me2 的结合较弱[11]。在酒酿酵母中过表达人NSD3S的PWWP结构域可诱导肿瘤代谢表型的获得,导致精氨酸水平显著下降,出现癌症中常见的代谢变化,首次建立了人类癌蛋白NSD3S和癌症代谢重编程之间的功能联系[12]。
PHD 锌指结构域,是一种可识别组蛋白甲基化的结构域[13]。在酵母菌中发现不同蛋白的PHD结构域可以分别识别H3K4me3或H3K36me3[14]。对NSD3的PHD5晶体结构研究发现,该结构域选择性结合未甲基化的H3K4和H3K9me3[15]。
2 NSD3在多种肿瘤中表达异常
组蛋白甲基化在染色质重塑和调控基因转录和细胞凋亡中起关键作用。NSD3的扩增或与其他基因融合等一直是许多肿瘤发生的重要特征,是重要的致癌基因(表1)。以下对NSD3与肿瘤之间的关系以及致瘤机制分别阐述。
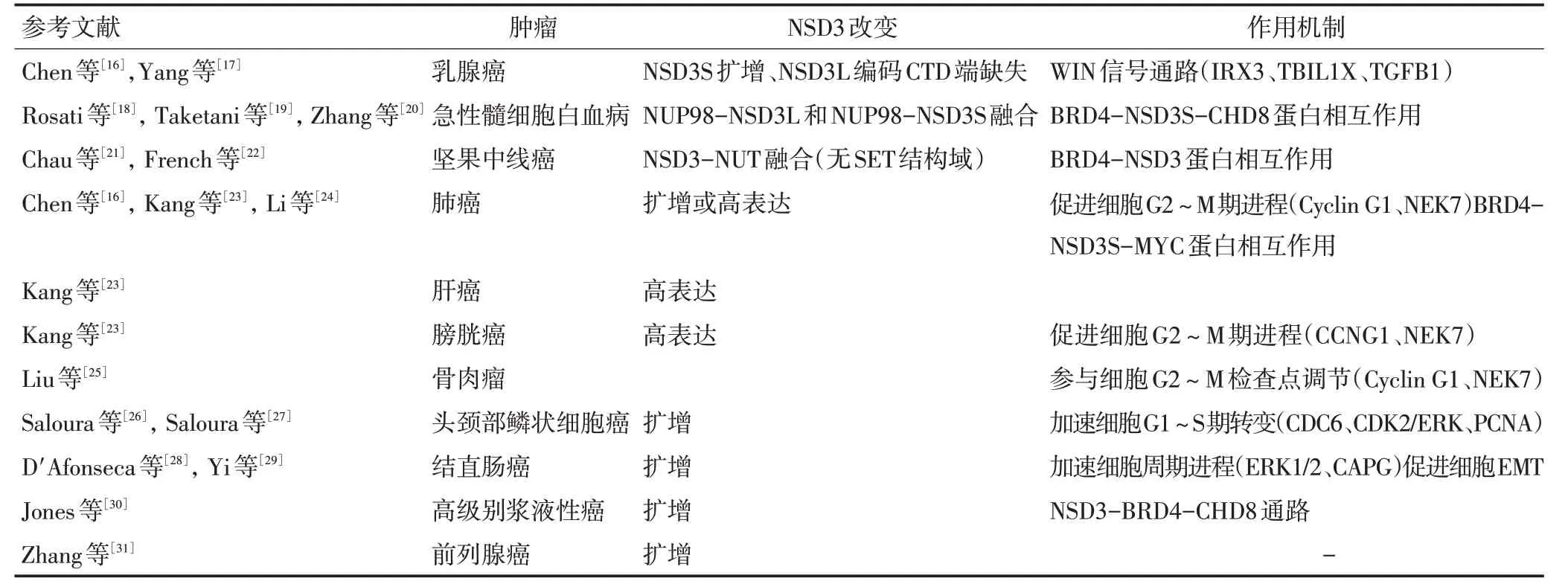
表1 与组蛋白甲基转移酶NSD3相关的肿瘤
2.1 NSD3与乳腺癌
约15%的原发性人类乳腺癌样本中发现了8p11-12 区域 的 扩增,FGFR1、LSM-1、C8orf4(TC-1)、RAB11FIP1、NSD3和ERLIN2是扩增区域中的候选癌基因,研究发现NSD3是所有8p11-12区癌基因中转化能力最强的。该区域癌基因的扩增与组织学分级和不良预后相关,同时免疫组织化学染色(immunohistochemistry,IHC)显示在原发性乳腺癌中NSD3 蛋白水平呈高表达[16-17]。将NSD3的基因通过慢病毒转导进入乳腺上皮细胞系MCF10A后,可使细胞在没有生长因子的情况下持续生长并形成扩张集落,过度表达NSD3的MCF10A细胞可形成明显异常的腺泡;相反,敲除NSD3基因可减慢细胞的增殖,并促进细胞死亡[17]。在8p11-12扩增的原发性乳腺癌标本中,基因组分析显示编码NSD3L的CTD片段(包括SET结构域)的基因区域出现缺失,而编码NSD3S的外显子1~10出现扩增[17]。NSD3可以上调IRX3(Iroquois homeobox 3)、下调TGFB1(trans⁃forming growth factor-beta 1)等目的基因的表达,从而可能通过活化WNT信号通路(Wnt/β-cantenin),促进肿瘤的形成[17]。
乳腺癌病毒启动子调控的转基因小鼠模型中,发现NSD3在乳腺上皮组织中高表达,导致导管上皮非典型增生,并最终形成原位癌和浸润性导管癌[32]。
2.2 NSD3与AML
在t(8;11)(p11;p15)易位的急性髓细胞白血病(acute myeloid leukemia,AML)患者中,发现核孔蛋白-98(nucleoporin 98 kDa,NUP98)-NSD3 融合转录本[18],同时在1例放射性相关骨髓增生异常综合征的原子弹幸存患者中,也发现了上述融合伴随del(1)(p22p32)缺失[19]。对该融合转录本的mRNA 进行分析显示,NUP98 的1~11 外显子与NSD3 的第4 个以及之后的外显子形成的框内融合转录本,可以形成NUP98-NSD3L和NUP98-NSD3S融合蛋白[19],具有该融合的AML预后差且易复发。
NSD3也可以与其他蛋白相互作用形成致癌复合物。在AML中,NSD3S可作为适配蛋白与含溴结构域蛋白4(bromodomain-containing protein 4,BRD4)和染色域-解旋酶-DNA结合蛋白8(chromodomain-helicase-DNAbinding protein 8,CHD8)结合,形成BRD4-NSD3S-CHD8相互作用,NSD3S在没有SET结构域的情况下,可以允许转录激活,是维持AML增殖的重要亚型,敲除NSD3基因可抑制人AML细胞系HL-60、MOLM-13[20,33]。
2.3 NSD3与NMC
坚果中线癌(NUT midline carcinoma,NMC)又称NUT carcinoma(NC)是一种带有NUT 融合基因的非常罕见且分化差的鳞状细胞癌。研究发现在141 例NMC 病例中NUT 融合基因比例分别为BRD4-NUT 78%,BRD3-NUT 15%,NSD3-NUT 6%,前者预后最差,后两者肿瘤转移率低于前者[21]。NSD3-NUT融合基因,由t(8;15)(p12;q15)易位形成,是NSD3外显子1~7 与NUT 外显子2~7 融合而形成的框内转录本。NUT 抗体免疫印迹验证了NSD3-NUT 融合癌蛋白的表达,该融合蛋白保留了N 端区PWWP 结构域但没有SET 结构域[22],该融合的第1~569 个氨基酸序列与NSD3S 相似,也间接表明NSD3S 在不具备甲基转移酶活性的情况下可以独立促进肿瘤的发生[22]。敲低NSD3-NUT使带有该融合基因的NMC细胞系1221 细胞系增殖减缓,分化增强[22]。BRD4/3-NUT 融合是由t(15;19)(q14;p13.1)易位引起的,该融合蛋白在转录因子参与下与RNA 聚合酶Ⅱ(RNA polymerase Ⅱ,RNA pol Ⅱ)结合,驱动致癌基因的表达以阻断肿瘤分化并推动肿瘤生长[34]。研究发现在BRD4-NUT 细胞系中敲低NSD3,或在NSD3-NUT 细胞系中敲低BRD4均可使相应细胞系增殖减缓,分化增强,说明BRD4可以与NSD3结合[22]。
2.4 NSD3与肺癌
使用组织芯片对肺癌组织进行检测,发现NSD3在癌细胞中的表达明显高于相应的非癌组织[23]。The Cancer Genome Atlas(TCGA)数据库挖掘与分析研究发现NSD3 在21%的肺鳞癌中扩增,NSD3 扩增的非小细胞肺癌细胞系H1581(7 个拷贝NSD3)和H1703(6个拷贝NSD3)中发现NSD3的mRNA和蛋白质水平在这些细胞系中呈高表达,其蛋白水平与NSD3 拷贝数呈正相关[16]。研究发现NSD3 SiRNA 沉默,降低了肿瘤细胞中H3K36me2 水平,从而影响了有丝分裂过程中的细胞周期蛋白G1(Cyclin G1)和NIMA 相关激酶7(NIMA related kinase 7,NEK7)的表达,使细胞停滞在G2~M期,G1期的癌细胞比例明显降低[23]。在肿瘤相关蛋白与蛋白相互作用分析实验中发现,在肺癌H1299 细胞系中,NSD3S 与MYC 结合,并且与BRD4形成BRD4-NSD3S-MYC相互作用,从而增加MYC 蛋白的稳定性以及MYC 癌蛋白目的基因的转录活性[24]。
2.5 NSD3与其他肿瘤
在膀胱癌和肝癌中,IHC证实NSD3高表达,敲低NSD3基因,可抑制膀胱癌细胞的增殖,降低了Cyclin G1期和NEK7的表达水平,使细胞停滞在G2~M期,G1 期的癌细胞比例明显降低[23]。随后在骨肉瘤中,发现NSD3可能参与G2~M期检查点的调节,抑制骨肉瘤细胞中的NSD3,会使处于G2~M期的细胞比例和凋亡细胞数量增加[25]。
NSD3 基因在头颈部鳞状细胞癌(squamous cell carcinoma of the head and neck,SCCHN)中出现扩增,并导致H3K36me2水平升高,通过上调与细胞周期蛋白依赖性激酶2(cyclin-dependent kinase 2,CDK2)促进细胞从G1期向S期转变,加速细胞增殖。NSD3扩增与该病的不良分级和重度吸烟史呈正相关[26]。该研究小组随后发现NSD3 可以单甲基化表皮生长因子受体(epidermal growth factor receptor,EGFR)酪氨酸激酶结构域中的赖氨酸K721,激活下游的细胞外信号调节激酶(extracellular signal-regulated kinase ERK)级联反应,增强其与增殖细胞核抗原(proliferat⁃ing cell nuclear antigen,PCNA)的相互作用,加速DNA复制,加快SCCHN细胞的周期进展[27]。
在结直肠癌中,对NSD 家族基因变异情况的研究发现,NSD3的基因扩增率最高,并且与其mRNA表达高度相关,符合肿瘤驱动基因的特点[28]。大肠癌中,NSD3过表达通过激活ERK1/2信号通路和增加肌动蛋白加帽蛋白(actin-capping protein CAPG)表达,刺激大肠癌细胞增殖、迁移和上皮-间充质转化(epi⁃thelial-mesenchymal transition,EMT)能力[29]。
TCGA 在输卵管、卵巢、子宫内膜来源的高级别浆液性癌的数据中发现存在NSD3-BRD4-CHD8 通路扩增,相比于无此通路扩增的肿瘤,预后更差,整体生存率更低[30]。TCGA 对399 例前列腺癌样本的51个HKMTases数据分析发现,发现雄激素受体高表达的癌组织中NSD3、SETD2、KMT2B、NSD1等出现明显扩增,并与前列腺癌Gleason评分显著相关[31]。
3 NSD3S与BRD4、C-MYC在肿瘤发生中的机制
BRD4是溴区结构域和末端外域(bromodomain and extraterminal,BET)蛋白家族成员之一,可以通过溴区结构域与乙酰化组蛋白结合,也可以通过磷酸化RNA pol Ⅱ来刺激转录延伸[35]。C-MYC蛋白(MYC)是一种基因表达调节因子,在细胞增殖、代谢、迁移和凋亡等细胞转化过程中起作用[24]。
研究发现BRD4存在以下几个转录激活特征:其CTD可直接募集阳性转录延伸因子b(positive transcriptional elongation factor b,P-TEFb),使RNA pol Ⅱ磷酸化刺激转录延伸[36];也可以通过ET结构域与NSD3S的第100~263氨基酸序列结合[37],后者第384~645氨基酸序列与CHD8结合,形成BRD4-NSD3S-CHD8相互作用,促进转录激活[33]。BRD4、NSD3S和CHD8在AML基因组中共定位,在使用BET抑制剂治疗时,它们可以从超级增强子区域释放出来[33]。研究发现NSD3与BRD4形成的复合物可调控H3K36的甲基化水平,使H3K36me3在转录活性区域富集,促进基因转录延伸,敲除或沉默NSD3,会导致H3K36me3水平降低,从而使基因转录停滞[37]。在NSD3-NUT融合的NMC细胞系(1221细胞系)中,NSD3的N端与BRD4的ET域结合,使用BET抑制剂,可诱导NMC细胞分化并抑制增殖[22]。
NSD3S与BRD4结合后,其PWWP结构域直接募集MYC,形成BRD4-NSD3S-MYC 相互作用。BRD4 在BRD4-NSD3-MYC途径中,通过独立的转录机制调节MYC;也可以通过BRD4-pTEFb介导的途径调节MYC,从而稳定MYC蛋白水平并增加转录活性[24],敲除NSD3S可导致MYC在mRNA和蛋白水平的表达下降。BRD4已证实可通过MYC启动子下游的超级增强子来调节AML细胞中MYC的表达[33]。NSD3S可干扰F-box和WD重复结构域(F-box and WD repeat domain containing 7,FBXW7)介导的蛋白酶体降解,来稳定MYC蛋白并增强其的半衰期和转录活性[38]。随后发现茶黄素3(theaflavin 3,30-digallate,TF-3),是NSD3/MYC相互作用的抑制剂,通过降低MYC的表达水平来抑制NSD3/MYC蛋白的相互作用,防止肿瘤的发生[39]。最新研究发现NSD3的PWWP1结构域特效拮抗剂BI-9321(甲基咪唑类),可特异性的针对该结构域的组蛋白赖氨酸结合位点,在AML细胞系MOLM-13和RN2中,可下调MYC基因的信使RNA的表达,从而抑制细胞的增殖,并增强BRD4抑制剂在MOLM-13细胞中的作用。这表明可使用BI-9321与BRD4拮抗剂联合抑制NSD3-PWWP1,为治疗与NSD3相关肿瘤提供一种新的治疗策略[40]。
4 展望
NSD3 在肿瘤的发生、发展中起重要作用并与肿瘤预后及复发密切相关,是目前新发现的转化型致癌基因。本文综合阐述NSD3 在肿瘤中的生物学功能,除了扩增过表达、局部缺失外还可以与NUP98、NUT、BRD4、CHD4以及MYC形成融合蛋白或相互作用,从而促进肿瘤周期进程,增加肿瘤侵袭能力。在NSD3 三种转录蛋白中以NSD3S 致癌作用最强,虽然只有一个PWWP 结构域,但能在不具备甲基转移酶活性的情况下独立促进肿瘤的发生,如NMC;而NSD3L 致癌作用却不如NSD3S;目前研究WHISTLE可以抑制基因转录,是抑癌蛋白,但机制尚不清楚。目前尚无针对NSD3 抑制剂,提示需要进一步对NSD3上、下游调控网络和结构特点进行研究,为今后肿瘤的临床治疗和预后提供有价值的指导。
猜你喜欢
热带作物学报(2022年7期)2022-08-06
保健与生活(2022年11期)2022-06-09
科学与生活(2021年16期)2021-11-25
湖南饲料(2021年4期)2021-10-13
保健与生活(2021年5期)2021-04-12
浙江医学(2020年19期)2020-10-20
陕西农业科学(2019年4期)2019-05-13
时尚育儿(2018年8期)2018-05-14
医学研究杂志(2015年9期)2015-07-01
癌变·畸变·突变(2015年4期)2015-02-27
