
大麦在网斑病菌侵染后的转录组学分析
2020-12-17 12:13汪军成姚立蓉李葆春马小乐司二静尚勋武王化俊孟亚雄
麦类作物学报 2020年9期
李 桃,汪军成,姚立蓉,李葆春,马小乐, 杨 柯,司二静,尚勋武,王化俊,孟亚雄
(1.甘肃农业大学农学院,甘肃兰州 730070; 2.甘肃省干旱生境作物学重点实验室/甘肃省作物遗传改良与种质 创新重点实验室,甘肃兰州 730070; 3.甘肃农业大学生命科学技术学院,甘肃兰州 730070)
栽培大麦(HordeumvulgareL.)是隶属禾本科的一年生作物,主要用途是饲用、啤酒酿造及食品加工等。由子囊菌亚门真菌网斑病菌(Pyrenophorateres,Ascomycotina)引起的大麦网斑病在国内大麦主要产区开始流行,并有逐年加重的趋势[1-2],其在世界其他国家如澳大利亚[3]、新西兰和丹麦[4]、南非[5]、美国[6]等地也均有发生。网斑病可抑制大麦生长,严重时会造成大麦减产、品质变劣,甚至会造成绝收,是限制大麦生产的主要原因之一。为解决网斑病对大麦的危害,大麦抗网斑病机制研究成为近年来被关注的重点,而通过基因组学方法鉴定和分离新基因是研究大麦抗病机制的有效方法之一。
有关大麦抗病基因分离和鉴定的报道并不多,且大多与大麦抗逆境胁迫有关,如HvNAC1[8]、Mlo[9]、ICS和PAL[10]等家族的基因。黄德华等[7]证实了大麦GER4基因簇的启动子表达与白粉病真菌病原体攻击有关,但有关大麦抗真菌类病害基因的研究并不多见。大麦基因组测序的完成[11]以及高通量测序技术的日益成熟,对大麦抗病基因的深入研究提供了方便。
植物的抗逆境胁迫不仅受多个信号传导途径的调控,还受多个基因协同表达和转录因子的调控[12]。有研究表明,与差异表达基因趋势相同的蛋白大多与植物抗逆相关;乙烯、赤霉素、苯丙烷类代谢参与甘蔗黑穗病侵染的响应过程[13]。因此,掌握关键基因的调控机制是实现促进多基因表达以提高植物综合抗病性的有效途径。研究发现,次生代谢产物[14]、植物激素信号转导[15]、MAPK信号传导[16]、鞘脂[17]等代谢途径在植物的抗病防御反应中发挥着重要作用,如NAC[8]、NF-Y[18]、HD-Zip[19-20]和WRKY[21]等家族的基因。为了解已知抗逆境基因是否与抗网斑病有关,本研究拟比较抗性和敏感型大麦材料对网斑病侵染后响应基因的表达特性,并通过比较其转录组(RNA-sep)探索大麦中有关抗病代谢途径,以期获得与抗病性有关的基因,了解大麦对网斑病菌侵染的响应机制,为大麦抗网斑病的进一步深入研究提供参考。
1 材料与方法
1.1 材料及设计
由甘肃农业大学麦类课题组提供抗病材料BYT-CYA3(B)和敏感型材料美41/I(M)。种子用72%乙醇处理 15 s,10%次氯酸钠处理 10 min,用蒸馏水冲洗6次后,置于铺有双层无菌滤纸的无菌培养皿里发芽,喷雾保持滤纸湿润;一周后,将发芽种子移栽入花盆。每盆种植5 株,每个品种共30盆。待幼苗长至两叶一芯期,对25盆大麦喷施由甘肃农业大学麦类课题组提供的活化网斑病病原菌菌株(LQA)悬浮液(浓度约106·mL-1),其余5盆为空白对照。接菌完成后置于21 ℃黑暗培养,于0、3、6、12、24和72 h后分别剪取接菌组与对照组叶片3~5 g,液氮速冻,于 -80 ℃保存备用。3次生物学重复。
1.2 RNA提取和cDNA 文库构建
采用Plant RNA Kit试剂盒(OMEGA,中国上海)按照说明提取大麦叶片总的RNA。在RNA样品中加DNaseI于 37 ℃水浴30 min,防止DNA污染。用超微量分光光度计检测总RNA的浓度。按照试剂盒说明富集mRNA和去除rRNA;用DNA探针杂交rRNA,再用RNaseH和DNaseI纯化RNA;用打断buffer使RNA片段化;反转录合成cDNA,组建cDNA文库。
1.3 转录组测序和数据过滤
在BGISEQ-500测序平台对构建好的cDNA文库进行双末端测序,所有测序工作由华大基因生物公司(深圳)完成。
测序所得的数据为原始数据(raw reads)。使用在线的SOAPnuke软件(https://github.com/BGI-flexlab/SOAPnuke)过滤原始数据,去除接头污染、未知碱基N过高的reads和质量低的reads,获得clean reads。利用在线软件HISAT(Hierarchical Indexing for Spliced Alignment of Transcripts)将clean reads与大麦参考基因组序列IBSC_PGSB_v2.39进行比对。使用 Bowtie2 (http://bowtie-bio.sourceforge.net/Bowtie2/index.shtml)将clean reads比对到参考基因序列得到比对结果[22],对比对结果使用RSEM(http://deweylab.biostat.wisc.edu/rsem/rsem-calculate-expression.html)[23]计算差异表达基因。分析转录组测序数据,包括转录本的覆盖度及分布。
1.4 基因的功能注释
将转录组测序鉴定到的所有基因合并为一个基因集数据库,使用getorf检测所有基因的 ORF;使用hmmsearch将ORF与转录因子蛋白结构域(数据来源于TF)进行比对,根据植物转录因子数据库(PlantTFDB)描述的转录因子家族特征对所有基因进行功能鉴定。使用HMMER程序的 hmmpfam与Pfam 23.0对转录因子与PlantTFDB进行比对,搜索蛋白结构域以鉴定编码转录因子的基因,使用DIAMOND(https://github.com/bbuchfink/diamond)软件将转录组测序鉴定的所有基因比对至植物抗病基因数据库(plant resistance gene database,PRGdb)进行注释,依据比对覆盖度(query coverage)和一致性(identity)等条件对注释结果进行筛选。对差异表达基因进行GO和KEGG功能注释富集分析(Wiki https://en.wikipedia.org/wiki/Hypergeometric_distribution)。本研究的转录组原始数据可以从NCBI数据库获取,登陆号SUB6851949。
1.5 网斑菌侵染大麦叶片响应的差异表达基因的鉴定和qRT-PCR验证
试验以接菌后0 h样品为对照(B-CK,M-CK),比较抗病和敏感型材料接种不同时间的样品(B-3h,M-3h,B-6h,M-6h,B-12h,M-12h,B-24h,M-24h,B-72h,M-72h)与相应对照材料基因的表达水平,参照Audic等[24]的方法,进行基因表达量分析,满足错配率[25](false discovery rate,FDR) ≤0.001且表达差异在2倍以上的基因为差异表达基因。
利用Primer-NCBI在线设计软件(https://www.ncbi.nlm.nih.gov/tools/primer-blast/#)设计引物(表1),对差异表达基因进行qRT-PCR验证分析。将样品3次生物学重复的 RNA 采用第1链cDNA合成试剂盒(TIANGEN,中国北京)按照说明合成反转录cDNA,以反转录cDNA为模板,以大麦Actin为内参,采用SYBR Green RT-PCR 试剂盒(TIANGEN),按照说明使用ABI 7500 Real Time PCR扩增仪进行荧光定量检测,结果按照2-ΔΔCT计算相对表达量。
2 结果与分析
2.1 测序结果质量评估
为了研究大麦在不同时间段响应网斑病菌侵染的转录组学变化特征,采集大麦接菌处理0 h、3 h、6 h、12 h、24 h和72 h的幼苗叶片,提取总RNA,构建了12个cDNA文库,用BGISEQ-500平台对其进行转录组学测序,平均每个文库测序产生10.63 Gb的数据,共获得1 401 716 667个raw reads,去除含接头的reads、未知碱基N含量大于5%的reads和低质量的reads,共得到 127 601 667条clean reads,过滤后12个cDNA文库的clean reads占raw reads的比例均在80%以上;clean reads的Q20比率均在97%以上,其 Q30比率均在86%以上,每个文库clean reads数均在101.61×106条以上(表2),获得的测序数据与质量符合下一步分析要求。
样品比对到基因组的平均比对率为 87.61%,比对到基因集的平均比对率为 74.93%;共检测到基因数为35 545个,其中已知基因30 030个,新基因为5 515个。

表1 实时荧光定量PCR所用的引物Table 1 Primers for real-time quantitative PCR
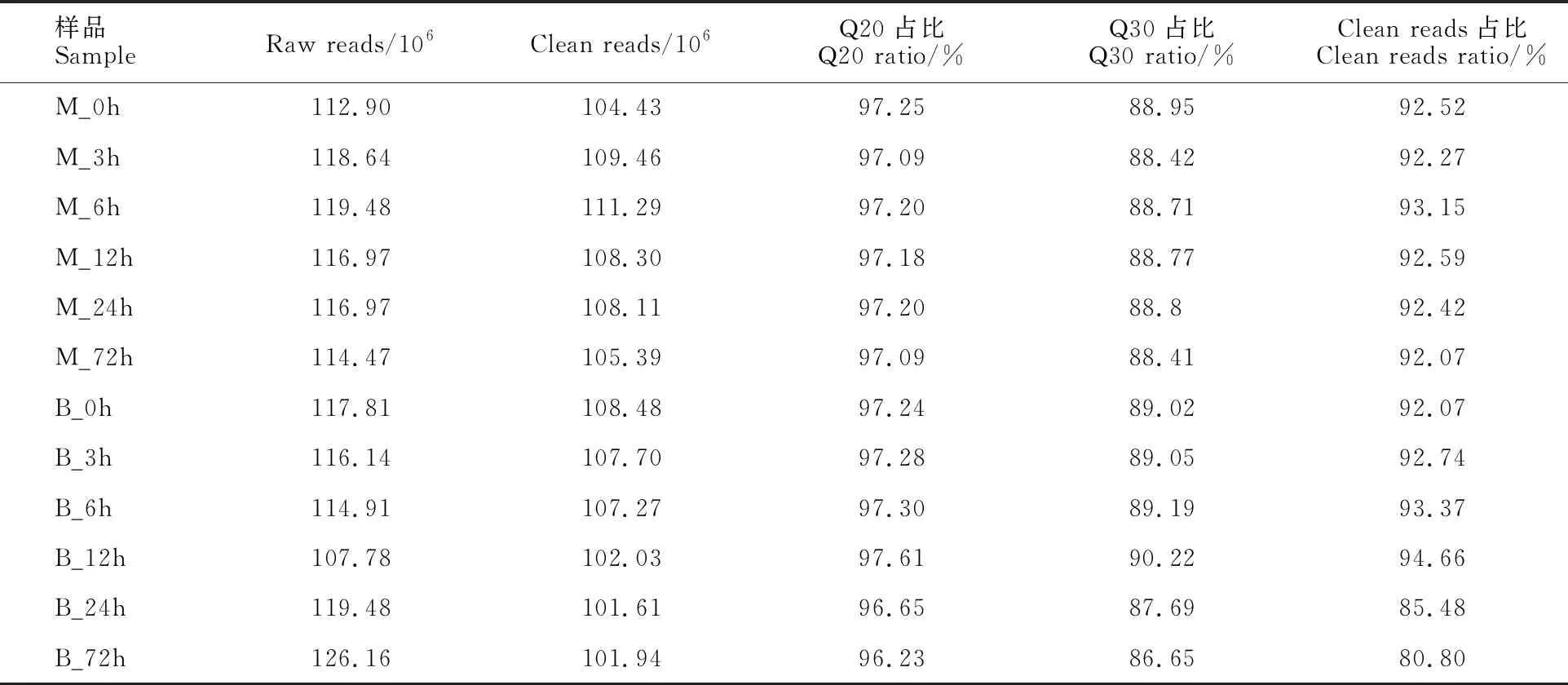
表2 供试材料Reads 的质量统计Table 2 Quality statistics of transcriptome sequencing after filtering
2.2 基因注释分析
对检测到的30 030个已知基因进行功能注释分析,发现只有1 977个基因在TF数据库有注释,可归类到58个基因家族,注释到基因数最多的是MYB家族(228),其次是AP2-EREBP(170)、bHLH(157)、FAR1(146)、NAC(145)、ABI3VP1(125)和WRKY家族(107),注释到基因数最少的有CAMTA(1)、HRT(1)、S1Fa-like(1)(图1 A)。在PRGdb数据库中,有3 427个基因被注释到13个结构域中,其中注释到RLP结构域中的基因数目最多,其次是NL、CNL、N和TNL,注释到的基因数分别有789、508、449和352个,注释到的基因数目最少的结构域是TNL-OT(图1 B)。

A:在TF数据库中的比对结果;B:在 PRGdb数据库中的比对结果。
2.3 网斑病菌侵染后差异表达基因(DEGs)分析
与网斑病菌侵染大麦0 h处理对照相比,在网斑病菌侵染大麦3 h、6 h、12 h、24 h和72 h后,抗病材料中分别有9 842、12 073、9 065、4 674和5 939个差异表达基因;敏感型材料中分别有10 149、11 322、11 142、2 305和5 023个差异表达基因。随着侵染时间的持续,抗病材料与敏感型材料上调和下调的差异表达基因(DEGs)数整体呈先增后减的趋势。抗病材料中的上调差异表达基因数目均大于敏感型材料(图2)。
2.4 差异表达基因的GO富集分析
对差异表达基因进行GO富集分析,发现这些差异表达基因在2种材料不同阶段的GO富集结果相似。在生物过程(biological process)中,差异表达基因主要富集的条目有生物调节(biological regulation)、细胞成分组织或生物发生(cellular component organization or biogenesis)、细胞过程(cellular process)、定位(localization)、代谢过程(metabolic process)、对刺激的反应(response to stimulus)。在细胞组分(cellular component)中,差异表达基因主要富集的条目为细胞(cell)、大分子复合物(macromolecular complex)、细胞膜(membrane)、细胞膜片段(membrane part)、细胞器(organelle)、细胞器的部分(organelle part)。在分子功能(molecular function)中,差异表达基因主要富集的条目有整合(binding)、催化活力(catalytic activity)、转运活动(transporter activity)。
在抗病材料BYT-CYA3中,网斑病菌侵染大麦接触阶段到侵入阶段(接种3~6 h),富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目均明显增加;在网斑病菌侵染的扩展阶段(接种12~24 h),富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目较侵入阶段均骤减;而在大麦发病阶段(接种72 h),与扩展阶段相比,富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目均明显增加。在敏感型材料美41/I中,从网斑病菌侵染大麦到扩展前期阶段(3~12 h),富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目没有明显变化;而从扩展后期到发病阶段,富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目均明显减少。

图2 不同时间的差异表达基因数Fig.2 Number of differentially expressed genes at different time
在网斑病菌侵染的接触阶段(3 h),抗病材料中富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目明显少于敏感型材料,其中,最为明显的条目有细胞成分组织或生物发生(cellular component organization or biogenesis)和细胞(cell)。在网斑病菌侵入阶段,抗病材料中富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目明显多于敏感型材料,最为明显的条目有细胞成分组织或生物发生(cellular component organization or biogenesis)、细胞过程(cellular process)、代谢过程(metabolic process)、细胞(cell)、细胞器(organelle)、细胞器的部分(organelle part)、整合(binding)、催化活力(catalytic activity)。在网斑病菌侵染大麦的扩展阶段(12~24 h),抗病材料中富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目明显少于敏感型材料,最为明显的条目有生物调节(biological regulation)、代谢过程(metabolic process)、细胞(cell)和整合(binding);在大麦发病阶段(72 h),抗病材料中富集到生物过程(biological_process)、细胞组分(cellular component)和分子功能(molecular function)三大功能条目中的差异表达基因数目均明显多于敏感型材料。
2.5 差异表达基因的KEGG富集分析
通过KEGG数据库对差异表达基因进行富集分析,发现这些差异表达基因在2种材料的不同阶段的KEGG富集结果比较相似。差异表达基因主要富集在12条通路上,即环境适应(environmental adaptation)、核苷酸代谢(nucleotide metabolism)、脂质代谢(lipid metabolism)、全球概览地图(global and overview maps)、碳水化合物代谢(carbohydrate metabolism)、其他次生代谢产物的生物合成(biosynthesis of other secondary metabolites)、氨基酸代谢(amino acid metabolism)、遗传翻译(translation)、转录(transcription)、折叠、分类和降解(folding,sorting and degradation)、信号转导(signal transduction)、运输和分解代谢(transport and catabolism)(图3)。
在抗病材料BYT-CYA3中,从网斑病菌侵染大麦接触阶段到侵入阶段(3~6 h),富集到各代谢通路上的差异表达基因数目均明显增加,最明显的通路有全球概览地图(global and overview maps)、碳水化合物代谢(carbohydrate metabolism)、其他次生代谢产物的生物合成(biosynthesis of other secondary metabolites)、氨基酸代谢(amino acid metabolism)、遗传翻译(translation)、转录(transcription)、折叠、分类和降解(folding,sorting and degradation)、信号转导(signal transduction)、运输和分解代谢(transport and catabolism);在网斑病菌侵染的扩展阶段(12~24 h),富集到各代谢通路上的差异表达基因的数目较侵入阶段均剧减;而在大麦的发病阶段(72 h),与扩展阶段相比,富集到各代谢通路上的差异表达基因数目均明显增加(图3 A)。在敏感型材料美41/I中,从网斑病菌侵染大麦接触阶段到侵入阶段(3 ~6 h),发现富集到各代谢通路上的差异表达基因数目均明显增加;而网斑病菌侵染的扩展阶段到大麦发病(12~72 h),富集到各代谢通路上的差异表达基因数目均较前阶段明显减少(图3 B)。
在网斑病菌侵染大麦的接触阶段(3 h),抗病材料中富集到各代谢通路上的差异表达基因多于敏感型材料,差异较大的代谢通路有转录(transcription)和全球概览地图(global and overview maps);从网斑病菌侵染大麦侵入阶段到扩展阶段(6~24 h),抗病材料和敏感型材料富集到各代谢通路上的差异表达基因数目均不断减少;在大麦发病阶段(72 h),抗病材料中富集到各代谢通路上的差异表达基因多于敏感型材料,差异较大的代谢通路有转录(transcription)和全球概览地图(global and overview maps)(图3)。
2.6 差异表达基因功能分析
在抗病材料中,共获得差异表达基因435个(图4A)。对435个差异表达基因进行KEGG途径富集分析,发现差异表达基因较多的通路有代谢途径(metabolic pathways;ko01100)、MAPK信号传导途径-植物(MAPK signaling pathway-plant;ko04016)、植物-病原体相互作用(plant-pathogen ;ko04626)、植物激素信号转导(plant hormone signal transduction;ko04075)和次生代谢产物的生物合成(biosynthesis of secondary metabolites;ko01110)(图4B)。其中有19.77%的差异表达差因基因通路产物是白介素-1受体相关激酶(interleukin-1 receptor-associated kinase)(图4B)。对435个差异表达基因进行GO途径富集分析,在生物过程(biological_process)中,差异表达基因主要富集的条目有细胞过程(cellular process)、代谢过程(metabolic process)。细胞组分(cellular component)中,主要富集的条目为细胞(cell)、细胞器(membrane)、细胞器的部分(membrane part)。在分子功能(molecular function)中,主要富集的条目为整合(binding)和催化活力(catalytic activity)(图4C)。推测这几条代谢通路与大麦抗病性有关。
为了分析差异表达基因的表达模式,对182个差异表达基因进行KEGG和GO途径分析,发现差异表达基因主要富集在过氧化物酶体(peroxisome;ko04146)、代谢途径(metabolic pathways;ko01100)、MAPK信号传导途径-植物(MAPK signaling pathway - plant;ko04016)、植物-病原体相互作用(plant-pathogen;ko04626)、植物激素信号转导(plant hormone signal transduction;ko04075)、代谢途径(biosynthesis of secondary metabolites;ko01110)。根据182个差异表达基因的功能在代谢通路上的定位,发现这些基因参与了维持细胞死亡的防御反应、维持病原体入侵和维持植物防御中活性氧的积累(图5B)。这182个差异基因可以作为大麦抗病基因进一步研究的候选基因。

A:网斑病菌侵染抗病材料的差异表达基因韦恩图分析;B:网斑病菌侵染抗病材料的差异表达基因的KEGG分类注释;C:表示网斑病菌侵染抗病材料的差异表达基因的GO分类注释。

灰框是通过转录组序列检测到的KEGG酶编码基因。
2.7 实时定量荧光PCR验证
为验证转录组测序的结果,从筛选的182个抗病相关基因中选取信号转导(signal transduction)、MAPK信号传导途径-植物(MAPK signaling pathway-plant)、其他次生代谢产物的生物合成(biosynthesis of other secondary metabolites)和植物-病原体相互作用(plant-pathogen)代谢通路的10个基因(表1),利用实时荧光定量PCR对其网斑病菌侵染前后的表达量进行分析,发现这10个基因的荧光定量与转录测序结果基本一致,且均上调表达。
3 讨 论
有研究表明,植物在与病原微生物的长期互作、协同进化过程中逐渐形成一系列的防卫机制,这些机制在诱导后才能快速、充分地表达,从组织结构机制、生理机制和分子机制看,与胁迫相关的基因在病原微生物侵染后的不同阶段具有不同的表达模式[26]。这现象可能是大麦调节体内生理代谢,以形成相应的防御机制。
本试验发现,大麦网斑病病菌侵染后无论是抗病材料还是敏感型材料,有大量不同家族的基因出现表达量的变化,两种材料在不同侵染阶段的差异表达基因的数目不同,这可能是其抗病性不同的根本原因。比较2个材料在网斑病菌侵染3 h、6 h、12 h、24 h和72 h后基因表达的异同,推测不同基因在网斑病菌侵染后,其转录水平调控方式在不同阶段存在差异。证明了王海波等[27]和翁 宇等[28]的研究结果:植物通过改变基因表达量来响应胁迫。
抗病材料和敏感型材料在病菌侵染后的差异表达基因有所相同,这表明不同抗病材料对逆境的响应机制不同。钱 前等[29]提出,植物有天然免疫抗性。本研究发现,即便在相同时间2个材料中存在的共同差异表达基因,其上调、下调表达程度也存在差异,推测这可能与大麦的基础抗性有关,同类型材料对相同胁迫有某种相同的响应机制,但响应程度不同。
本研究对差异表达基因做了PRGdb和TF注释,发现差异表达基因大多富集在CNL、N、NL、RLP和TNL等结构域中。有研究表明,富集在CNL、N、NL、RLP和TNL等结构域的基因与植物防御系统的抗病性有关[30-31]。目前,在植物中发现的与抗病性有关的基因家族有b ZIP、AP2/EREBP、WRKY、MYB、NAC等[32]。本研究中,差异表达基因在AP2/EREBP、NAC、WRKY和bHLH家族中分布数目较多。植物中特有转录因子家族是NAC,其参与植物对逆境胁迫的响应和,调控植物的抗病性[33-35]。王立国等[36]、董 帅等[8]和张立全等[37]完成了在陆地棉、大麦、黄花苜蓿中对NAC转录因子相关基因的克隆。证明了NAC参与调控植物的抗逆性。除了NAC,植物中还有一种特有的家族是锌指蛋家族WRKY。研究发现,WRKY类转录因子参与植物的形态发育与对病原体的防御响应[38]。目前,已有对WRKY基因的分析和结构域特点及其生物学功能的研究[39-40]和其在植物防御反应中的作用[41-42]方面的报道。本研究发现,MYB家族被注释到最多的基因数目。在植物中,MYB通过有效调控类黄酮物质的生物合成以调节机体免疫力、抗氧化、抗衰老及抵抗病毒能力[43]。已有研究表明,MYB与小麦对纹枯病、赤霉病抗性有关[44]。对于bHLH家族的研究表明,植物bHLH基因家族参与调控植物的生长和发育过程[45]。而对于AP2/EREBP家族的研究表明,在拟南芥中得到AP2/EREBP家族的独特成员是At4g13040基因,其参与SA介导的疾病防御1-APD1的Apetala 2家族蛋白合成,使病原体接种后无法诱导表达[46]。
本研究筛选出来182个可能与大麦抗病性有关的基因,其中,35个差异表达基因参与植物激素信号转导(plant hormone signal transduction;ko04075)代谢途径的基因(图3B),28个差异表达基因富集到信号转导(signal transduction),其中有7个差异表达基因参与合成LRR受体样丝氨酸/苏氨酸蛋白激酶FLS2(LRR receptor-like serine/threonine-protein kinase FLS2)(附表1)。苗丽丽等[47]研究发现,蔗糖非发酵相关蛋白激酶2(SnRK2)是植物特有的一类丝氨酸/苏氨酸蛋白激酶,SnRK2作为典型的多功能调节因子参与响应各种逆境胁迫;郭艳玲等[14]提出次生代谢产物参与植物的抗病防御反应,作为生化壁1垒防御病原物侵染,和信号物质共同参与植物的抗病反应。本研究中发现,45个差异表达基因参与其他次生代谢产物的生物合成(biosynthesis of other secondary metabolites)代谢途径的基因,其中9个差异表达基因参与合成过氧过氧化物酶体(peroxidase;K00430),而过氧化物酶是病原真菌抗氧化防御系统的主要组分[48]; 36个差异表达基因富集到环境适应(environmental adaptation),其中5个差异表达基因是抗病蛋白RPM1(disease resistance protein RPM1),8个差异表达基因参与合成LRR受体样丝氨酸/苏氨酸蛋白激酶FLS2(LRR receptor-like serine/threonine-protein kinase FLS2)。这些基因的结构与功能需要更多试验与探究。
本试验从分子水平上初步探索了大麦对网斑病侵染的响应机制,提供了抗病基因深入研究的候选基因,为大麦抗病基因的研究奠定基础。
猜你喜欢
今日农业(2022年4期)2022-06-01
当代水产(2022年1期)2022-04-26
中国农业科学(2021年23期)2022-01-14
休闲读品·天下(2021年2期)2021-10-08
疯狂英语·新读写(2021年2期)2021-02-25
中国瓜菜(2020年8期)2020-09-26
中国瓜菜(2019年8期)2019-09-19
发明与创新·大科技(2019年5期)2019-07-31
华人时刊(2016年19期)2016-04-05
中学生物学(2008年3期)2008-06-03
