
同位素内标-气相色谱-质谱法测定食用植物油中缩水甘油酯
2020-12-15 09:42:28张方圆王李平朱大林潘云山
中国油脂 2020年12期
张方圆,王李平,朱大林,潘云山
(广东省科学院 广东省测试分析研究所(中国广州分析测试中心),广东省化学危害应急检测技术重点实验室,广东省保健食品功效成分检测与风险物质快速筛查工程技术研究中心,广州 510070)
缩水甘油酯(Glycidyl esters,GEs)是缩水甘油和脂肪酸的酯化产物。GEs在人体中会代谢为缩水甘油(又称环氧丙醇),在2017年世界卫生组织国际癌症研究机构(IARC)公布的致癌物清单中,缩水甘油被列入了2A类致癌物清单。动物实验证明缩水甘油具有致癌作用,食用后容易对人体中枢神经产生抑制作用,可能损害生育能力甚至致癌[1]。另外,缩水甘油在氯离子和酸性条件下,易转换成氯丙醇,以3-氯-1,2-丙二醇(3-MCPD)为主,而3-MCPD 具有肾毒性、生殖毒性和致癌性[2],对人体健康产生巨大的威胁。
GEs已成为一种潜在的食品污染物,并引起了社会各界的广泛关注[3]。2016年,欧盟(EFSA)对GEs和氯丙醇酯类化合物对人类健康进行了风险评估,结果表明绝大多数植物油中均含有GEs和氯丙醇酯,并制定了氯丙醇的每日许可限量(TDI)值为0.8 μg/kg[4]。2018年,欧盟实施了对植物油和婴幼儿奶粉中GEs的限量管理[5]。目前,我国缺少食品中GEs的相关检测方法和标准,文献报道的方法主要分为直接法和间接法两类。直接法主要应用液相色谱-质谱法(LC-MS),在不破坏GEs结构的条件下,对各类GEs进行直接测定[6-7],但由于GEs种类较多,多种标准品还未商业化,导致标准品造价昂贵,且难以购得。间接法多采用气相色谱-质谱法(GC-MS),将GEs转化为其他特异性的物质,通过检测该物质的含量间接测定GEs的含量[8-9]。《2018年国家食品污染和有害因素风险监测工作手册》采用间接法测定GEs的含量,将GEs经酯交换反应后,与氯离子反应生成氯丙醇,通过氯丙醇的差值折算得到GEs的含量。间接法适用于测定GEs的总量,但由于GEs和氯丙醇酯的形成机理相似,氯丙醇酯通常伴随GEs一起形成,GEs不是氯丙醇形成的唯一前体,且食品中存在氯丙醇本底值的干扰情况[10]。因此,通过将GEs转化成氯丙醇间接测定GEs含量时,需先测定样品中氯丙醇及其酯的本底值,再测定样品中经GEs转化而来的氯丙醇和样品本身氯丙醇及其酯的总含量,通过计算氯丙醇含量的差值得到GEs的含量。这加大了实验工作量和实验所需样品量,且本底值测定的准确性会直接影响测定结果的准确性。
本研究采用间接法将GEs转化成3-溴-1,2-丙二醇(3-MBPD)酯,再通过水解和衍生化前处理方法,采用GC-MS测定3-MBPD含量,从而实现对食用植物油中GEs的测定,并通过正交实验对衍生条件进行优化。溴丙醇和氯丙醇结构和性质相似,且样品中不存在溴丙醇本底值干扰的情况,因此本方法可以通过测定溴丙醇含量直接得到GEs的含量,降低了样品中基质干扰,克服了氯丙醇本底值干扰,提高了检测的灵敏度和准确性,且减少了测定工作的样品需求量,可以更好地满足食用植物油中GEs含量的测定要求。
1 材料与方法
1.1 实验材料
1.1.1 原料与试剂
食用植物油样品均购于广州市各大超市;棕榈酸缩水甘油酯(GEs, 98.0%,BePure),棕榈酸-D5-3-氯-1,2-丙二醇酯(D5-3-MCPDE,100 μg/mL,First Standard);正己烷、甲基叔丁基醚、乙酸乙酯、丙酮、甲醇、三氯甲烷均为色谱纯;苯硼酸(PBA,97.0%,麦克林);实验用水为GB/T 6682—2008规定的一级水;其他试剂均为分析纯。
GEs标准储备液:准确称取棕榈酸缩水甘油酯标准品0.011 11 g(精确至0.000 01 g),用三氯甲烷配制成1 089 μg/mL的储备液,临用时用三氯甲烷稀释成所需质量浓度。
内标棕榈酸-D5-3-氯-1,2-丙二醇酯溶液:准确移取适量棕榈酸- D5-3-氯-1,2-丙二醇酯标准品,用乙酸乙酯配制成10 μg/mL的内标溶液。
1.1.2 仪器与设备
Agilent 7890A-5975C气相色谱-质谱联用仪,美国安捷伦科技有限公司;Sartorius BT125D 电子天平;TD25-WS多管架自动平衡离心机;KQ-600E超声波清洗仪;HH-4数显恒温水浴锅;0XW-80A旋涡混合器。
1.2 实验方法
1.2.1 样品前处理
称取植物油样品0.2 g(精确至0.000 1 g)于10 mL比色管中,准确加入20 μL内标溶液,再加入2 mL甲基叔丁基醚-乙酸乙酯溶液(体积比2∶1),涡旋溶解,作为待反应液。
向待反应液中加入酸化溴化钠溶液0.1 mL,涡旋混匀,在50℃水浴反应15 min;加入3 mL饱和碳酸氢钠溶液中止反应,并加入2 mL正己烷,涡旋,静置,取上清液于10 mL比色管中,用正己烷重复提取上清液2次,合并提取液于10 mL比色管中,40℃氮吹至近干,加入1 mL甲基叔丁基醚-乙酸乙酯溶液(体积比2∶1)复溶,备用。
于上述提取液中加入2 mL 1.8%硫酸-甲醇溶液,混合均匀后,置于40℃恒温水浴中反应16~20 h。加入3 mL饱和碳酸氢钠溶液中止反应后,涡旋,静置分层,氮吹除去有机溶剂。再用2 mL正己烷除去反应液中的脂溶物,弃上层液,正己烷重复脱脂2次,得到下层清液备用。
加入1.0 mL乙酸乙酯于上述清液中,涡旋提取1 min,取上层液于装有1 g无水硫酸钠的10 mL比色管中,重复提取2次,合并提取液,于40℃氮吹吹干。
加入适量PBA溶液,振荡均匀后,于恒温水浴反应一段时间,冷却,40℃氮吹至近干,加入1 mL正己烷,涡旋溶解,转移至进样小瓶中,待测定。
1.2.2 标准曲线的绘制
将GEs标准储备液稀释得到质量浓度为1.089 μg/mL标准溶液,分别移取GEs标准溶液20、50、100、200、500、1 000、2 000 μL于10 mL比色管中,并分别加入内标溶液20 μL,按1.2.1在酸性条件下与溴离子反应后,经1.8%硫酸-甲醇水解,乙酸乙酯提取,于40℃氮吹吹干后,按优化后的衍生条件衍生化,得到目标物3-MBPD-PBA质量浓度依次为0.021 8、0.054 4、0.109、0.218、0.544、1.089、2.178 μg/mL,内标质量浓度为0.200 μg/mL,然后进行GC-MS分析,以目标物峰面积与内标峰面积之比为纵坐标,3-MBPD-PBA质量浓度为横坐标绘制标准曲线。
1.2.3 GC-MS分析条件
GC条件:色谱柱为DB-5MS毛细管柱(30 m×250 μm×0.25 μm);进样口温度280℃;升温程序为60℃保持0 min,以6℃/min速率升温至180℃,再以60℃/min速率升温至300℃,保持3 min;进样方式为脉冲不分流;进样量1 μL。
MS条件:电子轰击离子源(EI);电离能量70 eV;离子源温度230℃;四级杆温度150℃,传输线温度280℃;扫描方式为选择离子扫描(SIM);溶剂延迟6 min;定量及定性离子见表1。

表1 3-MBPD-PBA、D5-3-MCPD-PBA的监测离子
2 结果与讨论
2.1 3-MBPD和D5-3-MCPD衍生物的定性分析
本实验中,GEs和内标D5-3-MCPDE在酸性条件下与溴离子反应后,经硫酸-甲醇水解后生成3-MBPD和D5-3-MCPD。3-MBPD和D5-3-MCPD在PBA用量100 μL、衍生温度50℃、衍生时间10 min条件下与PBA衍生后生成3-MBPD-PBA和D5-3-MCPD-PBA。衍生产物在GC-MS分析条件下,经全扫描模式分析后,得到相应的总离子流图和质谱图,如图1所示。
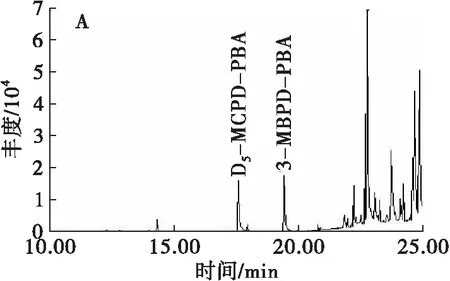
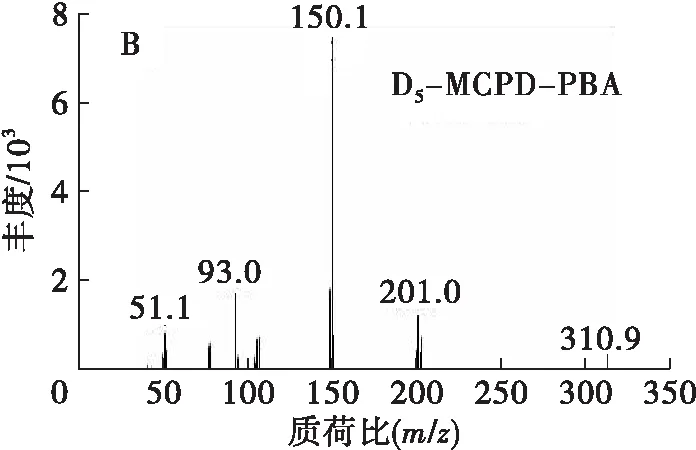
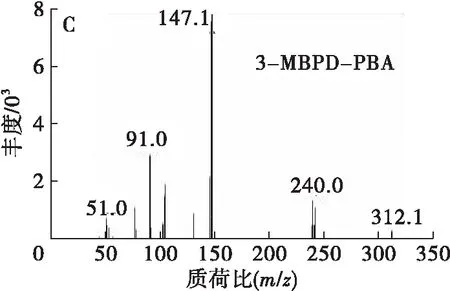
图1 2.178 μg/mL 3-MBPD、2.000 μg/mL D5-3-
从图1A可以看出,3-MBPD-PBA和D5-3-MCPD-PBA在该GC-MS分析条件下的出峰时间分别为19.4 min和17.6 min,两种物质的响应较好,且分离度良好。从图1B、图1C可以看出:D5-3-MCPD-PBA的特征离子为m/z150.1、201.0、93.0,其中m/z150.1的响应最高;3-MBPD-PBA 的特征离子为m/z147.1、240.0、91.0,其中m/z147.1的响应最高。因此,可采用选择离子监测模式对D5-3-MCPD-PBA和3-MBPD-PBA进行定性分析,以降低噪声和其他干扰效应,提高检测灵敏度。其中:D5-3-MCPD-PBA定量离子m/z150,定性离子m/z201、93;3-MBPD-PBA定量离子m/z147,定性离子m/z240、91。
2.2 衍生条件的优化
采用正交实验对3-MBPD与PBA的衍生反应条件进行优化。通过分析实验条件和前期单因素实验,在GEs质量浓度为21.78 μg/mL条件下,以PBA用量、衍生温度和衍生时间为因素,3-MBPD-PBA峰面积为指标进行正交实验设计。正交实验设计和结果见表2,方差分析见表3。
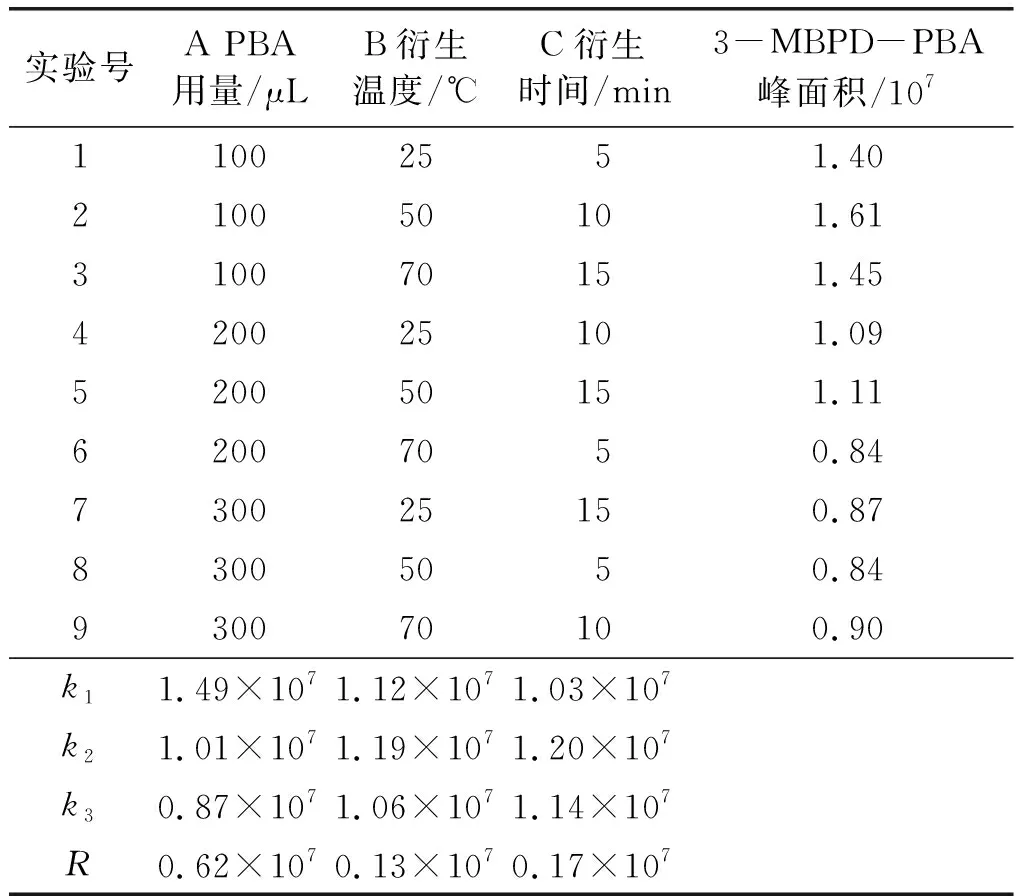
表2 正交实验设计和结果
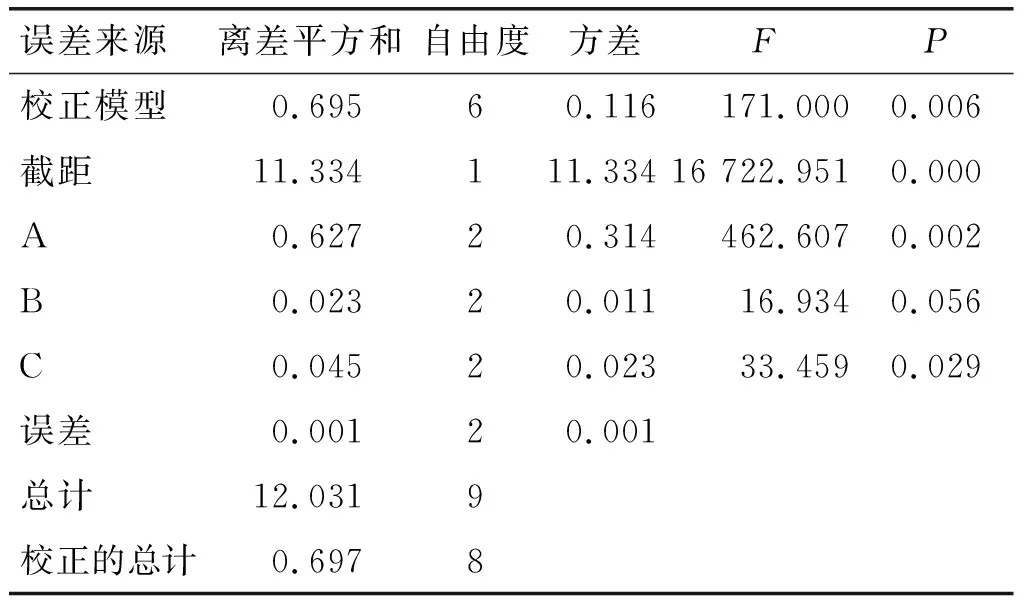
表3 方差分析
从表2可以看出,各因素对3-MBPD-PBA峰面积影响的主次顺序为A>C>B,最佳因素水平组合为A1B2C2,即PBA用量为100 μL,衍生温度为50℃,衍生时间为10 min。
从表3可以看出,各因素对3-MBPD-PBA峰面积影响的主次顺序为A>C>B,与极差分析结果一致。因素A和因素C对响应值有显著性影响(P<0.05)。
2.3 标准曲线与检出限
GEs在0.021 8~2.178 μg/mL的质量浓度范围内的标准工作曲线方程为y=4.255x-0.048 9,相关系数(R)为0.999 9。在该分析条件下,仪器信噪比为41,以3倍信噪比对应的质量浓度为检出限,可确定检出限为25 μg/kg。结果表明,本方法的线性关系良好,检出限能满足食用植物油中GEs的测定要求。
2.4 精密度和回收率
选取葵花籽油作为基质进行加标回收和精密度实验,考察方法的准确度和精密度。在3个不同水平上进行加标,分别进行6次平行实验,计算得到GEs的平均回收率和相对标准偏差(RSD),结果见表4。
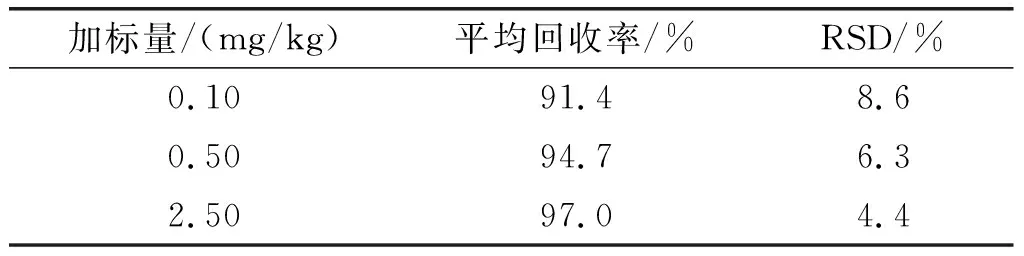
表4 基质加标回收率和精密度实验结果(n=6)
由表4可知,3个不同水平加标的平均回收率范围为91.4%~97.0%,RSD小于10%,准确度和精密度良好。这说明本实验所确定的前处理步骤和衍生化条件,适用于植物油中GEs的测定,测定结果准确、可靠,满足GB/T 27404—2008《实验室质量控制规范 食品理化检测》中检测方法确认的技术要求,可应用于植物油中GEs的测定。
2.5 样品测定
分别选取市售的10种食用植物油产品,按1.2.1、1.2.3方法进行GEs含量的测定,每个样品平行测定2次,取平均值,结果见表5。
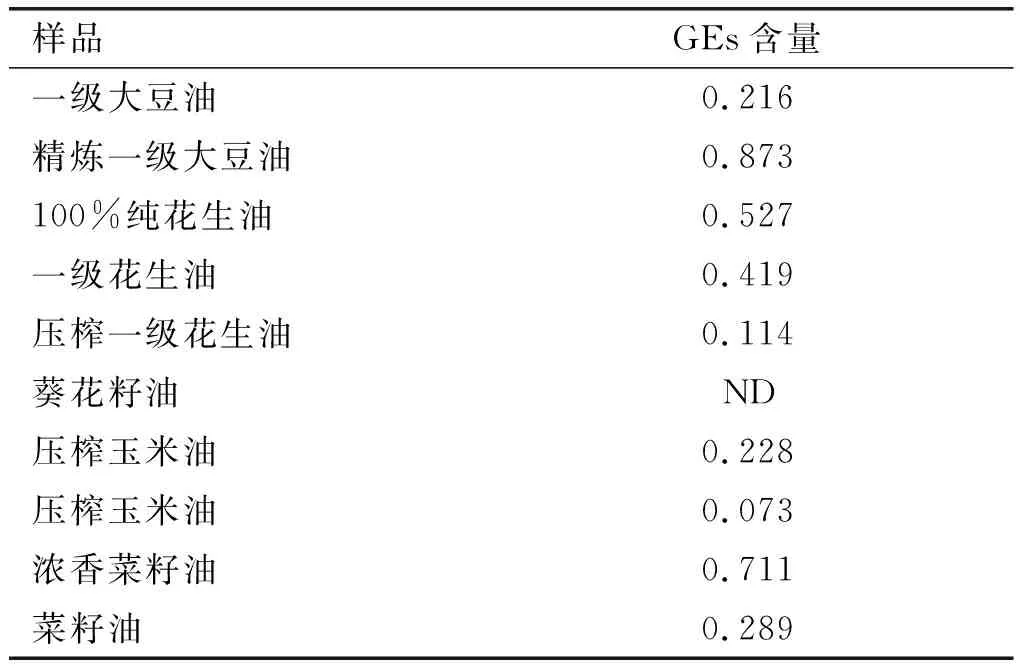
表5 10种食用植物油样品中GEs的含量 mg/kg
从表5可以看出,10个食用植物油样品中9个样品检出GEs,GEs的检出率高达90%,含量为0.073~0.873 mg/kg。欧盟EU 2018/290对植物油中GEs限量标准为1.0 mg/kg,参照欧盟限量标准可知,食用植物油中GEs的污染水平相对较高,存在一定的风险,这可能与食用植物油的生产过程和工艺有关,其中油脂精炼过程中的脱臭工艺是GEs产生的重要环节[11]。因此,建立准确可靠的测定方法用于监测食用植物油中GEs含量,控制GEs的污染风险,确保食用植物油的安全,是十分必要和紧迫的。
3 结 论
本文建立了同位素内标-气相色谱-质谱法用于食用植物油中GEs含量的测定。采用正交实验对衍生条件进行优化,确定3-MBPD与PBA的最佳衍生条件为GEs质量浓度为21.78 μg/mL时,PBA用量100 μL,衍生温度50℃,衍生时间10 min。本方法的检出限为25 μg/kg,平均加标回收率为91.4%~97.0%,RSD为4.4%~8.6%。方法可操作性强、重现性好,灵敏度、准确度和精密度均能满足实际工作中食用植物油中GEs的定性定量检测,可为食用植物油中GEs含量的检测标准制定和GEs风险监控提供重要依据。
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:30
石油炼制与化工(2022年2期)2022-02-15 11:42:26
食品工业科技(2021年23期)2021-12-16 02:21:50
食品安全导刊(2021年21期)2021-08-30 08:21:48
应用化工(2021年4期)2021-05-20 09:43:36
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
食品与生活(2017年5期)2017-05-27 20:28:38
农产品市场周刊(2017年6期)2017-03-10 20:34:29
合成化学(2015年10期)2016-01-17 08:56:48
