
甘蓝型油菜每角粒数的全基因组关联分析
2020-12-11 02:59:06孙程明斌张洁夫傅廷栋
作物学报 2020年1期
孙程明 陈 锋 陈 松 彭 琦 张 维 易 斌张洁夫,* 傅廷栋
研究简报
甘蓝型油菜每角粒数的全基因组关联分析
孙程明1,2陈 锋1陈 松1彭 琦1张 维1易 斌2,*张洁夫1,*傅廷栋2
1江苏省农业科学院经济作物研究所 / 农业部长江下游棉花与油菜重点实验室 / 江苏省现代作物生产协同创新中心, 江苏南京210014;2华中农业大学植物科学技术学院 / 作物遗传改良国家重点实验室, 湖北武汉 430070
每角粒数是油菜重要的产量构成因子, 增加每角粒数有助于提高油菜的籽粒产量。利用Illumina 60K SNP芯片对496份具有代表性的油菜资源进行基因型分析, 考察该群体在2个环境中的每角粒数, 利用MLM和GLM模型进行全基因组关联分析。结果表明, 本群体在2个环境中每角粒数的广义遗传力为57.7%。利用MLM和GLM模型分别检测到9个和20个位点, 所有MLM位点均得到GLM结果的验证。6个位点与前人定位的QTL重叠, 其中2个位点得到2次验证, 其余14个是新位点。在7个位点附近找到了候选基因, 其中在C09染色体的位点Bn-scaff_15576_1-p74980附近找到已克隆的油菜每角粒数基因, 在其余6个位点附近找到、等已知的拟南芥每角粒数基因的同源基因。本研究结果有助于解析油菜每角粒数的遗传基础及其调控机制, 为每角粒数的遗传改良奠定了基础。
甘蓝型油菜; 产量; 每角粒数; 关联分析; SNP标记
油菜是我国重要的油料作物, 种植面积约690万公顷,产油量占国产植物油总量的55%以上, 是我国最大的食用植物油来源[1]。作为食用油消费大国, 我国食用油60%以上却依赖进口, 油菜的产量水平直接影响我国食用油的供给, 因此, 提高产量成为油菜育种最迫切的目标。每角粒数是油菜重要的产量构成因子, 解析其遗传基础对提高油菜产量具有重要意义。
油菜种子的形成起始于胚珠原基, 而胚珠原基需要经历珠被、珠心和胚囊的形成, 以及授粉、受精、种子发育等过程才能最终形成种子, 这一系列过程受到复杂的遗传网络调控。李永鹏等[2]研究表明, 部分胚珠原基在发育过程中败育而最终不能形成成熟的种子, 正常发育胚珠百分比是反映品种间每角粒数差异的重要因素。Yang等[3]进一步研究表明, 油菜每角粒数的自然变异由每子房胚珠数(30.7%)、可育胚珠比(18.2%)、可育胚珠授精率(7.1%)和授精胚珠发育成种子的比率(43.9%)的差异共同决定。
多位研究者采用连锁分析定位了每角粒数QTL, Shi等[4]利用TN DH及F2群体在13条染色体检测到69个每角粒数QTL, 解释的表型变异范围是3.2%~15.5%; Luo等[5]利用60K SNP芯片再次对TN DH群体进行QTL定位, 共检测到64个每角粒数QTL, 其中贡献率最大的2个QTL和均位于A01染色体, 分别解释20.4%和24.1%的表型变异; 漆丽萍[6]在A01、A06~A08、C01、C03和C05染色体检测到15个QTL, 其中贡献率最大的2个QTL和均位于A01染色体, 分别解释26.3%和26.6%的表型变异; Cai等[7]在A01~A04、A06、A08~A10、C01、C03、C05、C06和C08染色体检测到17个QTL, 可解释5.6%~26.4%的表型变异; Yang等[8]利用一个RIL群体在A02、A06、C01、C04和C06染色体检测到5个QTL, 可解释5.3%~24.8%的表型变异, 其中位于A06的QTL在多个环境中被重复检测到, 解释20.1%的表型变异。
近年来, 随着高密度SNP基因分型芯片和全基因组测序等技术的发展, 全基因组关联分析(genome-wide association study, GWAS)已广泛应用于油菜复杂性状的遗传结构解析[9-12]。本研究以496份具有代表性的油菜资源为关联群体, 利用60K SNP芯片对群体进行基因型分析, 并对每角粒数进行全基因组关联分析, 旨在挖掘显著的SNP位点, 分析其候选基因, 为油菜每角粒数的遗传改良奠定基础。
1 材料与方法
1.1 试验材料
用于关联分析的496份甘蓝型油菜包括国内外的地方品种、育成品种及高世代育种材料(附表1)。其中国内资源444份, 主要来自湖北、重庆、江苏、湖南、四川、陕西等油菜主产省市; 国外资源52份, 主要来自德国、瑞典、朝鲜、加拿大等国家。所有材料均由华中农业大学国家油菜工程技术研究中心提供。
1.2 田间试验与表型调查
于2015年和2016年在江苏泰州(15TZ和16TZ)种植自然群体。采用完全随机区组设计, 设置2个重复, 单个材料每重复种2行, 每行15株, 行宽1.5 m, 行距40 cm。每年10月上旬大田直播, 田间管理按当地常规方式进行。在油菜成熟期, 取每小区8株长势一致的植株, 选主花序中部10个角果测量每角粒数。利用R软件对各环境的每角粒数表型进行统计分析和相关性分析, 并计算广义遗传力和最佳线性无偏预测值(best linear unbiased prediction, BLUP)[13-14]。
1.3 基因型检测与分析
利用Illumina 60K SNP芯片对496份甘蓝型油菜资源进行基因型分析。在Genome Studio软件中, 剔除Call Freq参数 < 0.75, 稀有等位基因频率 < 0.05, AA、BB频率 < 0.03及GenTrain值 < 0.50的SNP, 基因型杂合的SNP按缺失值处理。将过滤后的33,218个SNP序列比对至油菜Darmor-基因组, e-value阈值设为10-12, 保留19,167个单拷贝、位置清楚的SNP标记用于后续分析。
1.4 全基因组关联分析
用Structure 2.3.3进行群体结构Q矩阵的计算[15], 用SPAGeDi计算材料间的亲缘关系K矩阵[16]。将基因型、表型、Q矩阵和K矩阵导入Tassel 4.0后, 以Q矩阵和Q+K矩阵作协变量, 分别进行基于一般线性模型(General Linear Model, GLM)和混合线性模型(Mixed Linear Model, MLM)的全基因组关联分析[17]。参考前人文献[9-12], 关联分析的显著性阈值 (Bonferroni threshold) 定为1/总标记数, 即-lg() = 4.28。当1 Mb区间内存在多个显著SNP时, 如果两两间的2≥0.1, 则将这些SNP归为一个关联位点, 以最小值的SNP作为代表。用R软件包qqman绘制曼哈顿图和QQ (Quantile- Quantile)图[18]。
1.5 候选基因挖掘
以2= 0.1作为衰减阈值, 用Tassel计算显著关联位点的LD衰减距离作为其置信区间, 提取置信区间内的基因CDS序列, 与拟南芥的基因CDS进行BLAST比对, 设e-value阈值为10-10, 以相似度最高的拟南芥基因信息来注释油菜基因。将落在置信区间的油菜每角粒数基因和拟南芥每角粒数基因的同源基因作为候选基因。
2 结果与分析
2.1 每角粒数表型分析
496份油菜资源的每角粒数在2个环境中具有广泛的变异, 单个环境的每角粒数极差范围是15.24~18.90。单个环境表型平均值范围是(20.56±1.99)~(21.45±2.36), 变异系数范围是0.10~0.11 (表1和图1)。2个环境间的相关系数为0.55 (≤ 0.001), 达到极显著水平, 这表明本研究考察的表型数据具有较高的可靠性和重复性。每角粒数在2个环境的广义遗传力为57.7%。
2.2 每角粒数全基因组关联分析
利用MLM模型对各环境的每角粒数表型和BLUP值进行全基因组关联分析, 当阈值为−lg() = 4.28时, 在15TZ、16TZ和基于BLUP分别检测到6、2和2个显著位点(表2和图2)。位于C02的位点Bn-scaff_15712_6- p1336179在15TZ和16TZ两环境中被重复检测到。合并重叠的位点后得到9个显著位点, 分布在A01、A04、A07、A08、C01~C04染色体, 可解释3.26%~4.61%的表型变异。
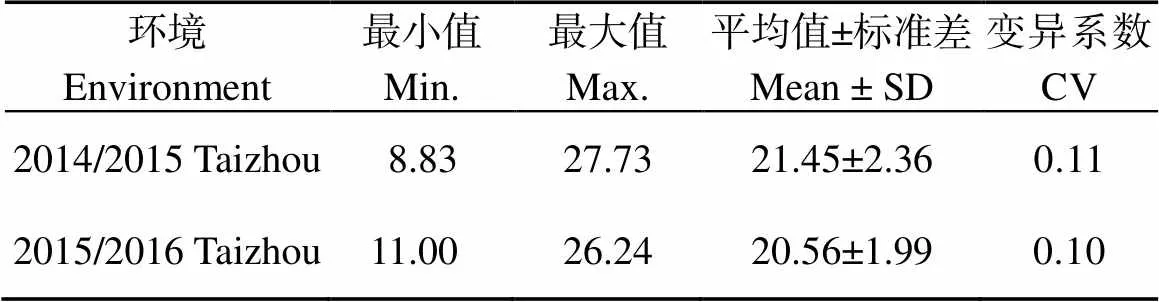
表1 关联群体每角粒数性状的统计分析

图1 关联群体在2个环境的每角粒数分布
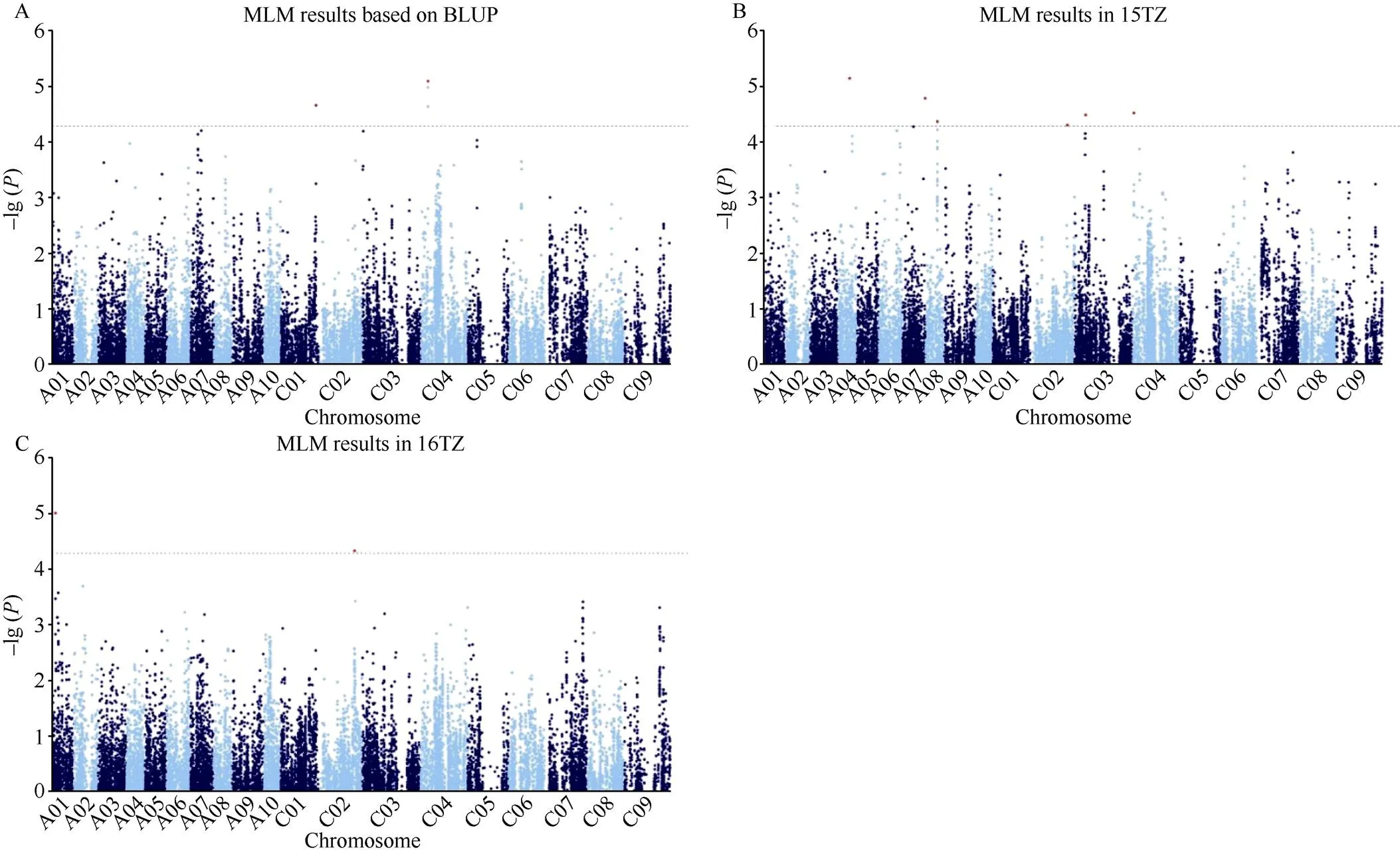
图2 油菜每角粒数全基因组关联分析(MLM)
A: 每角粒数BLUP值MLM曼哈顿图; B: 15TZ每角粒数MLM曼哈顿图; C: 16TZ每角粒数MLM曼哈顿图。水平线代表Bonferroni阈值。
A: Manhattan plot of MLM based on BLUP value; B: Manhattan plot of MLM in15TZ; C: Manhattan plot of MLM in 16TZ. The dashed horizontal line depicts the Bonferroni significance threshold.

表2 MLM每角粒数显著关联位点
利用GLM模型对各环境的每角粒数表型和BLUP值进行全基因组关联分析, 当阈值为−lg() = 4.28时, 在15TZ、16TZ和基于BLUP分别检测到10、6和5个显著位点。位于C04的位点Bn-scaff_23907_1-p3780基于BLUP和在15TZ均被重复检测到, 合并重叠的位点后得到20个显著位点, 分布在A01、A04、A07、A08、C01~C07和C09染色体, 可解释2.90%~4.83%的表型变异(表3和图3)。比较2个关联模型的结果, 所有MLM位点均得到GLM结果的验证, 合并2个模型的结果共得到20个显著位点。

图3 油菜每角粒数全基因组关联分析(GLM)
A: 每角粒数BLUP值GLM曼哈顿图; B: 15TZ每角粒数GLM曼哈顿图; C: 16TZ每角粒数GLM曼哈顿图。水平线代表Bonferroni阈值。
A: Manhattan plot of GLM based on BLUP value; B: Manhattan plot of GLM in 15TZ; C: Manhattan plot of GLM in 16TZ. The dashed horizontal line depicts the Bonferroni significance threshold.
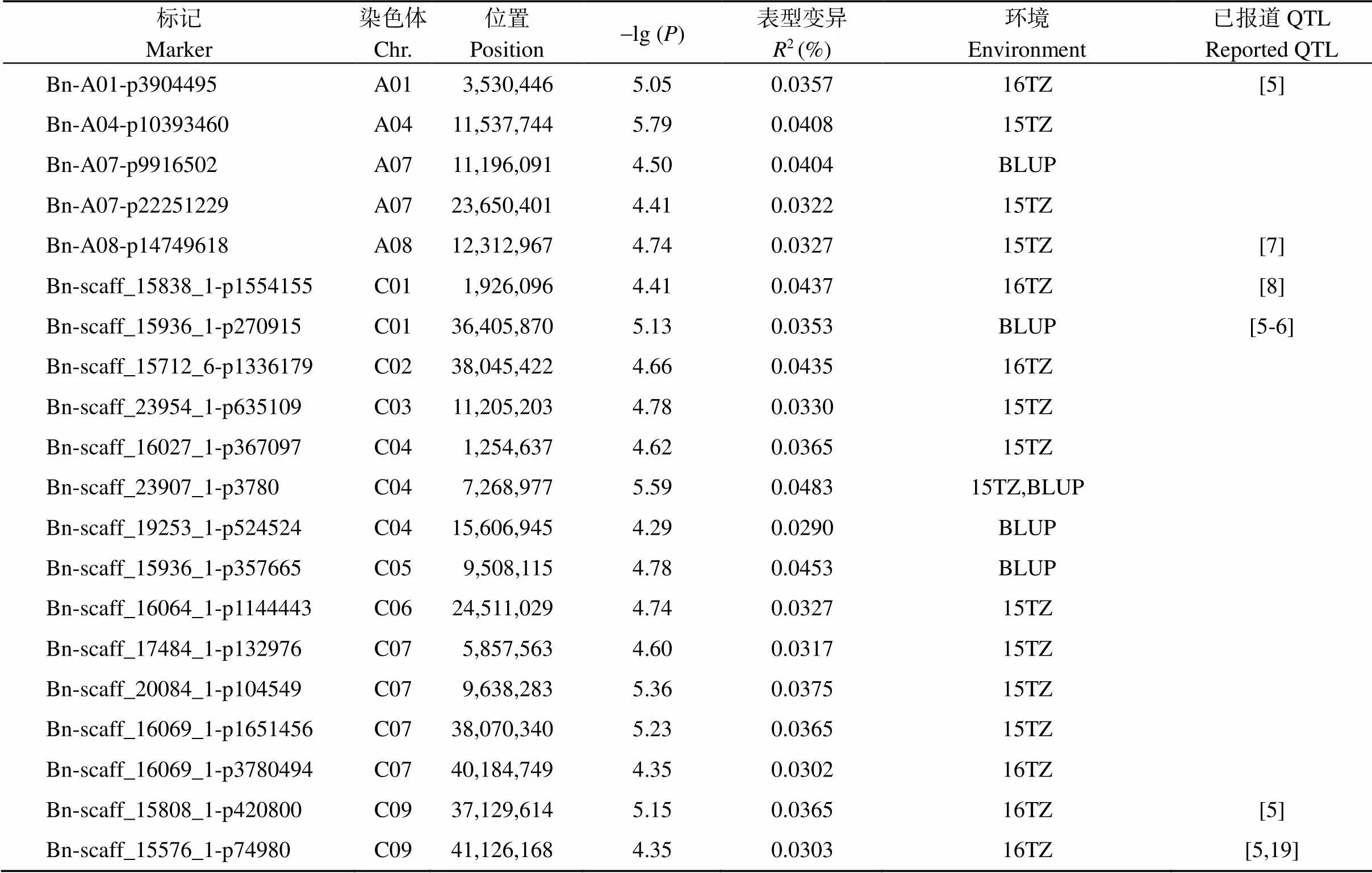
表3 GLM每角粒数显著关联位点
2.3 候选基因挖掘
基于油菜基因组注释信息, 在C09染色体Bn-scaff_ 15576_1-p74980位点下游82 kb找到已克隆的油菜每角粒数基因, 该基因是拟南芥的同源基因(表4)。参与调控油菜大孢子母细胞的减数分裂, 该基因的自然缺失导致部分雌配子体发育异常, 无法形成正常功能的胚珠, 最终导致油菜每角果粒数显著降低[19]。此外, 在A04染色体Bn-A04-p10393460位点上游522 kb找到候选基因, 该基因的拟南芥同源基因编码一个短富含甘氨酸结构域的蛋白, 该基因突变导致植株角果长度缩短、每角粒数减少、粒重降低[20]。在C03染色体Bn-scaff_23954_1-p635109位点下游162 kb找到候选基因, 该基因的拟南芥同源基因编码一个E3泛素连接酶, 其功能缺失突变体每角粒数增加、粒重增加[21]。此外, 本研究还找到其他候选基因, 包括拟南芥已知每角粒数调控基因[22]、[23][24]和[25]在油菜中的同源拷贝。
3 讨论
每角粒数是油菜重要的产量构成因子, 增加每角粒数有利于提高单株籽粒产量, 从而提高群体的籽粒总量。本研究对496份油菜资源的每角粒数进行2个环境的考察, 分析表明本群体的每角粒数广义遗传力为57.7%。利用2个关联模型MLM和GLM, 检测到20个显著位点, 分布于12条染色体。本研究中每角粒数关联位点的效应较小(< 5%), 在2个环境间能被重复检测到的位点少, 表明该群体中每角粒数主要受微效多基因调控, 且基因效应受环境影响较大。
本研究获得的每角粒数QTL有6个位点与前人结果一致(表2和表3), 其中位于C01染色体的位点Bn-scaff_15936_1-p270915, 物理位置36.41 Mb, 与Luo等[5]检测到的每角粒数QTL(范围: 36.53~37.48 Mb)和漆丽萍[6]检测到的QTL(范围: 35.69~36.81 Mb)重叠。位于C09染色体的位点Bn-scaff_15576_ 1-p74980物理位置是41.13 Mb, 与Li等[19]已克隆的每角粒数基因(位置: 41.21 Mb)和Luo等[5]检测到的QTL(范围: 41.28~41.67 Mb)重叠。这些结果证实了本研究结果的可靠性。此外, 本研究还检测到14个新位点, 如位于A04染色体的Bn-A04-p10393460和C04染色体的Bn-scaff_23907_1-p3780, 表型贡献率均相对较高; 位于C02染色体的位点Bn-scaff_15712_6- p1336179在2个环境中被重复检测到, 这些位点值得进一步验证和研究。
目前油菜中已报道的每角粒数基因较少, Li等[19]利用图位克隆的方法, 在C09染色体克隆了每角粒数基因; Yang等[26]应用CRISPR/Cas9系统敲除在油菜A04和C04染色体的2个拷贝, 双突变植株叶片增多, 每角粒数和粒重均增加; Shah等[27]发现拟南芥在油菜A02染色体的拷贝突变, 导致了油菜株高、分枝高度、分枝数、每角粒数和单株产量等性状的改变。本研究在Bn-scaff_15576_1-p74980位点下游82 kb找到(表4)。此外, 还在另外6个位点附近找到了候选基因, 这些基因的拟南芥同源基因均参与每角粒数的调控, 部分基因还有一因多效的作用, 如[20]和[22]同时影响角果长、粒型和粒重等性状。后续研究可通过转基因或CRISPR/Cas9敲除, 进一步验证这些候选基因的功能。

表4 每角粒数关联位点候选基因信息
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb.aspx。
[1] 王汉中. 我国油菜产业发展的历史回顾与展望. 中国油料作物学报, 2010, 32: 300–302. Wang H Z. Review and future development of rapeseed industry in China., 2010, 32: 300–302 (in Chinese with English abstract).
[2] 李永鹏, 程焱, 蔡光勤, 范楚川, 周永明. 油菜每角果粒数差异的细胞学基础和分子机理. 中国科学: 生命科学, 2014, 44: 822–831. Li Y P, Cheng Y, Cai G Q, Fan C C, Zhou Y M. Cytological basis and molecular mechanism of variation in number of seeds per pod in., 2014, 44: 822–831 (in Chinese with English abstract).
[3] Yang Y, Wang Y, Zhan J, Shi J, Wang X, Liu G, Wang H. Genetic and cytological analyses of the natural variation of seed number per pod in rapeseed (L.)., 2017, 8: 1890. doi: 10.3389/fpls.2017.01890.
[4] Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J. Unraveling the complex trait of crop yield with quantitative trait loci mapping in., 2009, 182: 851–861.
[5] Luo Z, Wang M, Long Y, Huang Y, Shi L, Zhang C, Liu X, Fitt B D, Xiang J, Mason A S. Incorporating pleiotropic quantitative trait loci in dissection of complex traits: seed yield in rapeseed as an example., 2017, 130: 1569–1585.
[6] 漆丽萍. 甘蓝型油菜株型与角果相关性状的QTL分析. 华中农业大学博士学位论文, 湖北武汉, 2014. Qi L P. QTL Analysis for the Traits Associated with Plant Architecture and Silique inL. PhD Dissertation of Huazhong Agricultural University, Wuhan, Hubei, China, 2014 (in Chinese with English abstract).
[7] Cai G, Yang Q, Chen H, Yang Q, Zhang C, Fan C, Zhou Y. Genetic dissection of plant architecture and yield-related traits in, 2016, 6: 21625. doi: 10.1038/srep 21625.
[8] Yang Y, Shi J, Wang X, Liu G, Wang H. Genetic architecture and mechanism of seed number per pod in rapeseed: elucidated through linkage and near-isogenic line analysis., 2016, 6: 24124. doi: 10.1038/srep24124.
[9] Sun C, Wang B, Yan L, Hu K, Liu S, Zhou Y, Guan C, Zhang Z, Li J, Zhang J, Chen S, Wen J, Ma C, Tu J, Shen J, Fu T, Yi B. Genome-wide association study provides insight into the genetic control of plant height in rapeseed (L.)., 2016, 7: 1102. doi: 10.3389/fpls.2016.01102.
[10] Lu K, Wei L, Li X, Wang Y, Wu J, Liu M, Zhang C, Chen Z, Xiao Z, Jian H. Whole-genome resequencing revealsorigin and genetic loci involved in its improvement., 2019, 10: 1154. doi: 10.1038/s41467-019-09134-9.
[11] Chen L, Wan H, Qian J, Guo J, Sun C, Wen J, Yi B, Ma C, Tu J, Song L. Genome-wide association study of cadmium accumulation at the seedling stage in rapeseed (L.)., 2018, 9: 375. doi: 10.3389/fpls.2018.00375.
[12] Xu L, Hu K, Zhang Z, Guan C, Chen S, Hua W, Li J, Wen J, Yi B, Shen J. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (L.)., 2015, 23: 43–52.
[13] Merk H L, Yarnes S C, Van Deynze A, Tong N, Menda N, Mueller L A, Mutschler M A, Loewen S A, Myers J R, Francis D M. Trait diversity and potential for selection indices based on variation among regionally adapted processing tomato germplasm., 2012, 137: 427–437.
[14] Ihaka R, Gentleman R. R: a language for data analysis and graphics., 1996, 5: 299–314.
[15] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study., 2005, 14: 2611–2620.
[16] Hardy O J, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels., 2002, 2: 618–620.
[17] Bradbury P J, Zhang Z, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 2633–2635.
[18] Turner S D. qqman: an R package for visualizing GWAS results using QQ and manhattan plots., 2014, 1: 005165. doi: http://dx.doi.org/10.1101/005165.
[19] Li S, Chen L, Zhang L, Li X, Liu Y, Wu Z, Dong F, Wan L, Liu K, Hong D. BnaC9. SMG7b functions as a positive regulator of the number of seeds per silique inby regulating the formation of functional female gametophytes., 2015, 169: 2744–2760.
[20] Rodríguez-Hernández A A, Muro-Medina C V, Ramírez-Alonso J I, Jiménez-Bremont J F. Modification of AtGRDP1 gene expression affects silique and seed development in., 2017, 486: 252–256.
[21] Xia T, Li N, Dumenil J, Li J, Kamenski A, Bevan M W, Gao F, Li Y. The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis., 2013, 25: 3347–3359.
[22] Braud C, Zheng W, Xiao W. LONO1 encoding a nucleoporin is required for embryogenesis and seed viability in Arabidopsis., 2012, 160: 823–836.
[23] Groszmann M, Paicu T, Smyth D R. Functional domains of SPATULA, a bHLH transcription factor involved in carpel and fruit development in Arabidopsis., 2008, 55: 40–52.
[24] Chen N N, Chen H R, Yeh S Y, Vittore G, Ho H D. Autophagy is enhanced and floral development is impaired in AtHVA22d RNA interference Arabidopsis., 2009, 149: 1679–1689.
[25] Leroy O, Hennig L, Breuninger H, Laux T, Köhler C. Polycomb group proteins function in the female gametophyte to determine seed development in plants., 2007, 134: 3639–3648.
[26] Yang Y, Zhu K, Li H, Han S, Meng Q, Khan S U, Fan C, Xie K, Zhou Y. Precise editing of CLAVATA genes inL. regulates multilocular silique development., 2018, 16: 1322–1335.
[27] Shah S, Karunarathna N L, Jung C, Emrani N. An APETALA1 ortholog affects plant architecture and seed yield component in oilseed rape (L.)., 2018, 18: 380. doi: 10.1186/s12870-018-1606-9.
Genome-wide association study of seed number per silique in rapeseed (L.)
SUN Cheng-Ming1,2, CHEN Feng1, CHEN Song1, PENG Qi1, ZHANG Wei1, YI Bin2,*, ZHANG Jie-Fu1,*, and FU Ting-Dong2
1Institute of Industrial Crops, Jiangsu Academy of Agricultural Sciences / Key Laboratory of Cotton and Rapeseed (Nanjing), Ministry of Agriculture / Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing 210014, Jiangsu, China;2National Key Laboratory of Crop Genetic Improvement / College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, Hubei, China
Seed number per silique (SSN) is a key component of seed yield in rapeseed, increasing SSN can improve the seed yield of plants. A collection of 496 representative rapeseed accessions was genotyped by the Illumina 60K SNP array and phenotyped for SSN in two environments. The genome-wide association study (GWAS) of SSN was performed via the MLM (Mixed linear model) and GLM (General linear model). The broad-sense heritability of SSN was 57.7%. Nine and twenty loci were detected with MLM and GLM, respectively, and all loci detected by MLM were included those by GLM. Six loci were overlapped with reported QTLs, and two of them were validated by two independent researches, and the rest 14 loci were new. We identified plausible candidate genes nearby seven loci, and the reported rapeseed SSN genewas found near the locus Bn-scaff_15576_1-p74980 on C09 chromosome detected in this study. Besides, six candidates orthologous to documentedSSN genes, like,,,and, were found near our GWAS loci. The results provide an insight into the genetic basis of seed number per silique and lay a foundation for further mechanism exploration and breeding for this trait in.
L.; yield; seed number per silique; GWAS; SNP
2019-04-15;
2019-08-09;
2019-09-11.
10.3724/SP.J.1006.2020.94060
本研究由国家重点研发计划项目(2018YFD0100602), 国家现代农业产业技术体系建设专项(CARS-12), 江苏省农业科技自主创新基金(CX(19)3055), 江苏省基础研究计划(自然科学基金)项目(BK20190260)和中央高校基本科研业务费专项资金资助项(2662016PY063)资助。
The study was supported by the National Key Research and Development Program of China (2018YFD0100602), the Earmarked Fund for China Agriculture Research System (CARS-12), the Jiangsu Agriculture Science and Technology Innovation Fund (CX(19)3055), the Natural Fund Project of Jiangsu Basic Research Program (BK20190260), and the Fundamental Research Funds for the Central Universities (2662016PY063).
张洁夫, E-mail: jiefu_z@163.com; 易斌, E-mail:yibin@mail.hzau.edu.cn
E-mail: suncm8331537@gmail.com
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20190910.1542.018.htm
猜你喜欢
烟草科技(2022年9期)2022-09-24 08:58:58
今日农业(2021年21期)2021-11-26 05:07:00
今日农业(2021年14期)2021-10-14 08:35:40
山西农业科学(2021年4期)2021-04-19 08:51:56
心声歌刊(2019年4期)2019-09-18 01:15:28
山西农业科学(2018年7期)2018-07-13 06:00:40
现代园艺(2017年21期)2018-01-03 06:41:32
数学小灵通(1-2年级)(2017年10期)2017-11-08 08:39:47
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
