
Is vitamin D receptor a druggable target for non-alcoholic steatohepatitis?
2020-12-10 04:08YingCaoXiangBingShuZeminYaoGuangJiLiZhang
World Journal of Gastroenterology 2020年38期
Ying Cao, Xiang-Bing Shu, Zemin Yao, Guang Ji, Li Zhang
Abstract Nonalcoholic steatohepatitis (NASH) is a progressed stage of non-alcoholic fatty liver disease, and available therapeutic strategies for NASH are limited. Vitamin D receptor (VDR) is proposed as a druggable target for NASH due to the discovery of vitamin D deficiency in NASH patients. To date, vitamin D supplementation has not consistently conferred expected therapeutic benefits, raising the question of whether VDR can serve as a proper drug target for NASH. It is known that VDR can interact with other ligands such as bile acids in addition to vitamin D, and its expression can be induced by fatty acids, and insulin. It has also been shown that while activation of VDR in hepatic macrophages and hepatic stellate cells resulted in attenuation of hepatic inflammation and fibrosis, activation of VDR in hepatocytes could accelerate lipid accumulation. Thus, the multiplicity of VDR ligands, together with the cell type-specificity of VDR activation, must be taken into consideration in assessing the validity of VDR being a potential druggable target for NASH treatment. To this end, we have evaluated the relationship between VDR activation and various contributing factors, such as gut microbiota, bile acid, fatty acids, and insulin, in addition to vitamin D, with an expectation that a potential drug might be identified that can elicit VDR activation in a tissue- and/or cell type-specific manner and therefore achieving therapeutic benefits in NASH.
Key Words: Vitamin D receptor; Non-alcoholic steatohepatitis; Vitamin D; Bile acids; Inflammation; Lipid metabolism
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is characterized by excessive lipid accumulation in the liver. Recently, NAFLD has been redefined as “metabolic associated fatty liver disease” because of its close association with obesity, type 2 diabetes mellitus, and cardiovascular disease that are common features of metabolic syndromes[1-4]. Clinically, NAFLD presents a spectrum of symptoms ranging from nonalcoholic fatty liver (NAFL)and non-alcoholic steatohepatitis (NASH), to the related liver fibrosis and cirrhosis. NAFLD has become the most common liver disease worldwide, with an incidence rate of approximately 25%[5], and an average incidence rate of 7.6% in children[6]. Although in most cases NAFL is asymptomatic with no overt clinical manifestation, approximately 20%-30% of NAFL patients develop into progressive NASH. It has been estimated that NASH will become the most common indication for liver transplantation by the year 2030[7].
To date, pathogenic mechanisms underlying the development or progression of NASH remain unclear. Theories hypothesizing a “two-hits” or “multiple parallel hits” mechanism for NASH pathogenesis have been proposed, postulating the involvement of both hepatic and non-hepatic factors in the disease progression, including insulin resistance, oxidative stress, endoplasmic reticulum stress, bile acid metabolism, adipokines and gut microbiota[8-10]. In addition, genetic polymorphisms as well as epigenetic factors might also play a role in NASH development. Lifestyle modification is the first-line recommendation for NASH; thus even a modest (10%) yet sustained weight loss can confer a significant improvement in NASH and liver fibrosis[11]. In spite of lifestyle improvement, pharmacological interventions are desirable. Currently, several pre-clinical trials (phase II and III) aiming at bioenergetics, lipotoxicity, inflammation, and/or fibrogenesis are ongoing. Thus far, none of the trials have achieved desirable outcome and thus no Food and Drug Administration approved drugs are available.
Vitamin D is a lipid-soluble vitamin and its metabolite 1,25-dihydroxy-vitamin D3 [1,25(OH)2D3] functions as a steroid hormoneviabinding to vitamin D receptor (VDR). While vitamin D is mostly reported to participate in calcium/phosphate metabolism and bone homeostasis[12,13], recent studies have shown that lowered vitamin D levels are present in NAFLD/NASH patients[14-16]. Deficiency in vitamin D might result in upregulation of inflammation and oxidative stress genes in the liver (through endotoxin and toll-like receptor pathways), that are hallmarks of NASH[17,18]. Thus, attempts have been made to retard NASH progression with vitamin D supplementation.
VITAMIN D SUPPLEMENTATION IN THE TREATMENT OF NAFLD
Vitamins are small organic molecules with catalytic effects vital to health. There are six known subtypes of vitamin D with varied side chains, ranging from vitamin D2 to D7[12]. The most important vitamin subtypes in humans are D3 (cholecalciferol) and D2 (ergocalciferol). Vitamin D2 mainly comes from plants and present in fortified foods, whereas vitamin D3 is formed in the skin. Provitamin D (7-dehydrocholesterol) is converted into pre-vitamin D3 in the skin, upon exposure to ultraviolet B radiation at wavelengths between 290 and 315 nm[13]. The resultant pre-vitamin D3 is transported to the liver, where it is hydroxylated at C25 to form 25 hydroxyvitamin D3 [25(OH)D3], the major form of circulating vitamin D clinically used as a biomarker in monitoring vitamin D status. In the kidney, 25(OH)D3 is further hydroxylated at the C1 position of the A ring to form 1,25(OH)2D3, which is the most biologically active form of vitamin D.
Rodent studies have shown that prophylactic vitamin D treatment could reduce liver fat accumulation, relieve liver and intestinal inflammation, decrease the expression of fibrogenic genes, and retard the development of NASH[19,20]. In light of clinical observations that lowered level of circulating 25(OH)D3 was present in NASH patients[21-23], these animal studies provide tantalizing evidence that vitamin D supplementation might be of therapeutic value in the treatment of NASH. In a doubleblinded, randomized, placebo-controlled trial, vitamin D3 treatment (50000 IU for 12 wk) resulted in improved homeostasis model assessment-insulin resistance, serum alanine transaminase (ALT), aspartate transaminase, C-reactive protein, and adiponectin, but with no effect on body weight or blood lipids[24]. In a parallel, doubleblind, placebo-controlled study where NAFLD patients were randomly allocated to receiving 50000 IU vitamin D3 (n= 27) or placebo (n= 26) for 4 mo, patients receiving D3 showed significant decrease in serum malondialdehyde, indicative of attenuated oxidative stress[25]. Another prospective study showed that in 40 NASH patients with confirmed vitamin D deficiency, treatment with 20000 IU vitamin D per week for 4 wk resulted in marked improvement in hepatic steatosis[26]. These animal and clinical studies argue favorably the benefit of vitamin D supplement in ameliorating NAFL and/or NASH progression.
Conflicting results, however, have been reported about the benefit of vitamin D supplement in the management of NAFL or NASH. In a study of NASH patients with vitamin D deficiency, vitamin D3 supplementation (2100 IE for 48 wk) showed little improvement in hepatic steatosis, even though its effect in reducing plasma ALT level was significant[27]. In another prospective study, 13 patients pathologically diagnosed with NASH subjected to vitamin D3 supplementation (25 000 IU/week for 24 wk) showed no change in liver biochemistry, insulin resistance index or adipocytokine profiles before and after treatment, nor was there any change in liver histology (examined by liver biopsy) before and after a high-dose of vitamin D3 treatment[28]. More concerning was a report that patients with NAFLD developed vitamin D intolerance[29]. Thus, clinical evidence for and against vitamin D supplement are inconsistent, which may stem from the lack of clearly defined mechanisms underlying the vitamin D action.
EXPRESSION OF VDR IN THE LIVER
VDR is ubiquitously expressed with highest expression in gastrointestinal tract and endocrine tissues (https://www.proteinatlas.org/ENSG00000111424-VDR/tissue). Perhaps because hepatic expression of VDR is not robust, the physiological role of VDR in the liver was ignored for some times[30]. Nevertheless, expression of VDR is detectable in rodent livers with a context-specific expression pattern. Thus, while hepatic parenchymal cells (i.e., hepatocytes) exhibited low level of the VDR mRNA, non-parenchymal cells, such as sinusoidal endothelial cells, resident macrophages [i.e., Kupffer cells (KCs)], and hepatic stellate cells (HSC), expressed high levels of the VDR mRNA in rats[31]. Likewise, the VDR mRNA was readily detectable in hepatocytes, KCs, cholangiocytes, and stellates isolated from mouse livers, among them KCs showed the highest VDR expression[32]. The functional significance of VDR expression in these non-parenchymal cells in the rodent livers is largely unknown.
Varied level of VDR expression has also been noted in different stages of liver pathology (e.g., NAFLDvsNASH). Examinations of human liver biopsy samples obtained from patients with NASH or chronic hepatitis C have detected VDR signals in parenchymal cells as well as inflammatory cells. The intensity of VDR signal in cholangiocytes was inversely related to the severity of steatosis, lobular inflammation, and the NAFLD activity score in the NASH patients[33]An inverse relationship between the liver VDR expression and the severity of liver fibrosis and inflammation was also observed in patients with chronic hepatitis C[34]. The liver VDR expression was up-regulated in patients with NAFLD, and VDR induction is more significant in patients with simple steatosis as compared to that in NASH[33]. A decrease in liver VDR expression was also observed in a diet-induced NASH mouse model[35]. These results may suggest an upregulation of VDR expression at early stages of NAFLD, which is followed by a downregulation of VDR expression in the steatohepatitis stage.
DIFFERENTIAL ACTIVATION OF VDR IN THE LIVER
The nuclear receptor VDR is composed of six functional domains, of which the E domain (encoded by exon V and exon IX of the VDR gene) is responsible for ligand binding. The VDR not only binds to its canonical ligand vitamin D, but also binds to other ligands including secondary bile acids, especially lithocholic acid (LCA) and its metabolites (3-kitoLCA, 6-kitoLCA,etc.)[36]. After entering the cells, the VDR ligands are translocated into the nucleus, and the translocation induces VDR phosphorylation. The phosphorylated VDR subsequently forms a heterodimer with retinol X receptor (RXR), and the resultant VDR-RXR heterodimer promotes binding of VDR to vitamin-D-responsive element (VDRE) located in the promoter regions of the effector genes[37]. The VDR-RXR thus acts as a molecular switch that transduces the signal of VDR ligands to VDRE, and VDR ligands in turn stabilize VDR-RXR conformation[38].
"Endogenous” 1,25(OH)2D3 synthesis and VDR activation in the liver macrophages
The VDR ligand 1,25(OH)2D3 originates from two synthetic pathways, one is derived from 25(OH)D3 hydroxylation in the kidney, and the other is synthesized in activated macrophages. The 25(OH)D3 hydroxylation pathway starts in the liver where previtamin D3 is hydroxylated to form 25(OH)D3 (also known as calcidiol or calcifediol), catalyzed by several cytochrome P450 isoforms (e.g., CYP27A, CYP2R1, CYP3A4, and CYP2J3, among which CYP2R1 is the most relevant). The resultant 25(OH)D3 is converted into 1,25(OH)2D3 in the kidney through another hydroxylation reaction catalyzed by CYP27B1. The biologically active product 1,25(OH)2D3 is released into circulation and distributed to other cells. In the case of 1,25(OH)2D3 synthesis in immune cells (e.g., tissue resident macrophages), expression of CYP27B1 is markedly increased in activated immune cells, leading to augmented synthesis of 1,25(OH)2D3[39-43]. This resident macrophage synthesized 1,25(OH)2D3 serves as the “local ligand” of VDR within the immune cells and exerts its function endogenously[40]. In light of the critical role of liver macrophages during inflammation and injury, it is probably not outside the realm of possibility that exposure of VDR in stressed liver macrophages with endogenous 1,25(OH)2D3 (in addition to circulating 1,25(OH)2D3) could lead to context-specific activation.
Bile acid metabolism and VDR activation in hepatocytes
Bile acids, synthesized in the liver as cholesterol (steroid) derivative, play a major role in food digestion and absorption in the intestine. The majority of bile acids are reabsorbed from the distal ileum, and the remainders enter the colon and converted into secondary bile acids by the action of gut microbiomes. A mixture of bile acids (primary, secondary, conjugated, non-conjugated) in the colon can also be reabsorbed into the liver through portal vein. Excretion and reabsorption of bile acids are the two main components of a process termed “enterohepatic circulation,” which drives bile flow, eliminates certain lipids, endogenous metabolites (e.g., bilirubin) and toxins in the liver, facilitates absorption of lipids and lipid-soluble vitamins, and optimizes the bacteria flora in the intestine[44]. Bile acids also possess functions resembling those of hormones in modulating metabolic, endocrine and immune responses by way of signaling through their respective receptor molecules. The currently known bile acid receptor proteins include farnesoid X receptor (FXR), G protein coupled receptor, pregnane X receptor (PXR), and notably, VDR[36,45].
The ability of bile acids to activate different seemingly unrelated nuclear factors highlights the multiplicity of these hormone-like steroid derivatives. The nature of this multiplicity in bile acid binding to various nuclear factors is not fully elucidated. It is known that primary bile acids (e.g., CDCA and CA) can act as a ligand of the FXR and PXR. However, neither PXR nor FXR responds to 1,25(OH)2D3, the canonical ligand of VDR. On the other hand, VDR can be effectively activated by bile acids such as LCA, glycol-LCA, and keto-LCAs, and it has been shown that LCA and 3-keto-LCA compete effectively with 1,25(OH)2D3 for binding to VDR[36]. The mechanisms underlying the bile acid- or vitamin D-driven VDR activation may be different. Thus, it is reported that while vitamin D-driven VDR activation was associated with an increase in calcium levels, bile acid-driven VDR activation was not related to calcium flux[46].
The enterohepatic circulation of the hormone-like bile acids may contribute to VDR activation in the liver and intestine. In cholestasis, LCA level is increased in the liver and intestine, and the accumulation of LCA activates VDR, which converts LCA into a less toxic intermediate for excretion[47]. It has been shown that LCA can induce CYP3A4 expression in the liver through VDR, thus participating in bile acid oxidation and mediating cell detoxification[48]. Deficiency in intestinal VDR was associated with an increase in LCA-induced liver necrosis, whereas ectopic expression of CYP3A4 in an intestine-specific VDR breakage mice (VDRΔIEpC) resulted in an attenuation in LCAinduced hepatotoxicity, probably through inhibition of a bile acid transporters[49]. Studies have shown that in rats deficient in vitamin D, LCA treatment could increase serum calcium levels through VDR, in addition, LCA treatment resulted in increased expression of VDR and enhanced anti-inflammatory functions[50,51]. The increased expression of VDR upon LCA treatment was associated with activation of SIRT1/Nrf2 pathway, thus reducing NF-κB phosphorylation and IL-8 secretion[52,53]. Moreover, the ligand-activated VDR expression may be tissue-specific; thus while 1,25(OH)2D3 preferentially activates VDR in the upper intestine, LCA activates VDR in the lower intestine[46].
Bacterial metabolites also play a role in VDR function. Bacterial metabolites, such as butyrate, increases expression of intestinal VDR and inhibits the inflammatory response in mice[54]. The VDR-knockout mice showed altered intestinal microflora, with a significant decrease in lactic acid-producing bacteria, and an increase inclostridiumandbacteroidesin comparison of wildtype mice[55]. These data suggest an interconnection between VDR and intestinal microbiomes. Since microbiome metabolites can enter the liver through portal vein, the liver cells are exposed to various VDR ligands, which may variably affect the outcome of vitamin D supplementation in the treatment of NAFLD.
DIFFERENTIAL VDR ACTIVATION IN SUBSETS OF LIVER CELLS
The VDR homologous members, such as FXR, PXR, and constitutive androstane receptor (CAR), affect almost all aspects of the liver functions, including glycolipid metabolism and bile acid homeostasis. Because of its low expression in hepatic parenchymal cells, the functional significance of VDR in the liver has been less appreciated[56]. Nevertheless, the level of VDR expression varied widely across different regions and different cell types within the liver, thus regional activation of VDR is most likely of cell type-specific significance.
Liver macrophages are able to produce 1,25(OH)2D3 from its precursor, 25(OH)D3, and the resultant 1,25(OH)2D3 can induce macrophage differentiation through a VDRdependent mechanism[57].In vitrostudies have shown that 1,25(OH)2D3treatment could lead to increased phagocytic activity of macrophages, accompanied by increased secretion of antimicrobial peptides, cathelicidin, and defensin B2/4[58,59]. VDR activation in macrophages has also been shown to induce a strong immunosuppressive effect by inhibiting MHC class II antigens that are involved in antigen presentation[58]. Thus, activation of VDR in resident macrophages has been shown to prevent liver inflammation, steatosis, and insulin resistance in mouse liver, and protect the liver from endoplasmic reticulum stress[32,60].
HSCs activation is generally believed to be the initial stage in the progression of liver fibrosis. Activation of the VDR pathway has been shown to antagonize transforming growth factorβ/SMAD-dependent transcriptional responses of multiple pro-fibrotic genes in HSC[61]. Activation of VDR in HSC was also associated with an enhanced binding to P62 (a component of Mallory-Denk Bodies and Hyaline granules)/SQSTM 1, thus probably contributing to the retarded progression of liver fibrosis and liver cancer[62].
The VDR protein and VDR mRNA are detectable in human hepatocarcinoma HepG2 cells as well as in human primary hepatocytes. It has been reported that VDR in hepatocytes might play a role in inhibiting bile acid synthesis, thus protecting liver from injury in cholestasis[47]. Transcriptomic and metabolomic analyses of hepatocytes transfected with human VDR have revealed that 20% of the VDR responsive genes were related to metabolism of lipids (including glycerolipids and phospholipids), uptake of fatty acids, betaine, and glycerol, suggesting a critical role of VDR in lipid metabolism in cultured hepatocytes[63-65]. In a global VDR-deficient mouse model, the livers were protected from steatosis, suggesting that VDR-mediated processes may contribute to NAFLD development[66], either due to enhanced lipid synthesis or reduced lipid turnover, or both. In hepatic-specific VDR knockout mice (generated by crossing VDRflox/+with albumin-Cre mice), however, the livers became more susceptible to steatosis by a high-fat diet[67]. Thus, some contributing factors, other than VDR alone, must play a role in regulating hepatic lipid metabolism under high-fat diet conditions.
One such a contributing factor is insulin resistance. Insulin resistance is a common feature associated with metabolic syndromes, including NASH. Often, insulin resistance presents hyperinsulinemia, which is linked to unbridled lipolysis leading to increased fatty acid flux into the liver and, as a consequence, augmented hepatic lipogenesis[68]. It is known that hepatic VDR expression is induced by fatty acids and insulin[64]. Thus, the influence of hyperinsulinemia on hepatic expression of VDR and its potential relationship with hepatic steatosis under high-fat diet condition requires further investigation. In addition, the possibility of any unknown side effects caused by long-term vitamin D supplementation to the liver must be carefully monitored.
CONCLUSION
Besides the critical role in regulating calcium and phosphate metabolism, VDR may also play a role in the development and progression of NASH. Since vitamin D is not the sole VDR ligand, studies with vitamin D supplementation may not adequately reflect the true action of VDR in disease progression or prevention. The controversies about the efficacy of vitamin D supplement derived from many clinical investigations may stem from two oversights, one is the contribution of non-vitamin D ligands (such as secondary bile acids) to VDR activation, and the other is cell type heterogeneity of the liver (e.g., parenchymal cellsvsnon-parenchymal cells such as macrophages and HSC). The liver resident macrophages, when activated, have the capacity to synthesize biologically active vitamin D locally. This “endogenous” vitamin D, along with kidney-derived 1,25(OH)2D3 and other ligands in the circulation, can conceivably activate VDRin situand modulate immune responses. Activation of VDR in HSCs is also associated with an anti-fibrogenesis process. The low level expression of VDR in parenchymal cells renders the cell insensitive to its ligands (e.g.bile acids and circulating 1,25(OH)2D3). However, activation of parenchymal VDR may lead to lipid accumulation in the liver (Figure 1).
Since VDR exerts a rather diverse range of effects on different cells, the current strategies that non-specifically activate VDR through vitamin D supplementation may not be proper. In addition, long-term high dose vitamin D supplementation might cause other unexpected effects. Secondary bile acids (LCA, keto-LCAs, glycol-LCA) are potentially potent ligands for hepatocyte VDR activation, and gut microbiota plays a fundamental role in the production of LCA and its metabolites. These contributing factors need to be taken into considerations in the assessment of VDR activation (or attenuation) in therapies. It is likely that activation of VDR in non-parenchymal cells can protect the liver from overwhelming immune response, whereas activation of parenchymal VDR may bring undesirable outcomes.
Physiologically, liver immune cells are quiescent with low expression of CYP27B1 and VDR, and hepatocytes are exposed to low levels of VDR ligands. The metabolic circuits in different cell types may be interconnected in forming a delicate network to maintain and support the metabolic homeostasis within the liver as well with other parts of the body. NASH patients, although often in the vitamin D deficient status, are not unusual in the situation of excessive bile acids, fatty acids, and insulin. While the non-parenchymal cells might synthesize “endogenous” 1,25(OH)2D3 through a compensatory mechanism to activate VDRin situ, parenchymal cells can alternatively activate VDR by non-vitamin D ligands such as LCAs. Immune cells may synthesize relatively limited 1,25(OH)2D3 to activate VDR since the process is under the control of liver 25(OH)D3 provision. However, hepatocytes are continuously exposed to VDR ligands, and may accelerate lipid accumulation in the liver under metabolically compromised conditions (e.g., hyperlipidemia and hyperinsulinemia).
There is, therefore, a need to understand fully the mechanisms by which VDR is activated in a cell type-specific fashion and the associated pathophysiological consequences upon VDR activation at different stages of disease development. Tissuespecific and cell type-specific VDR agonists or antagonists are expected, which can then selectively activate (or inactivate) VDR-mediated process in precision. Until then, more in-depth studies to define the regulatory mechanisms of VDR in NASH are merited.
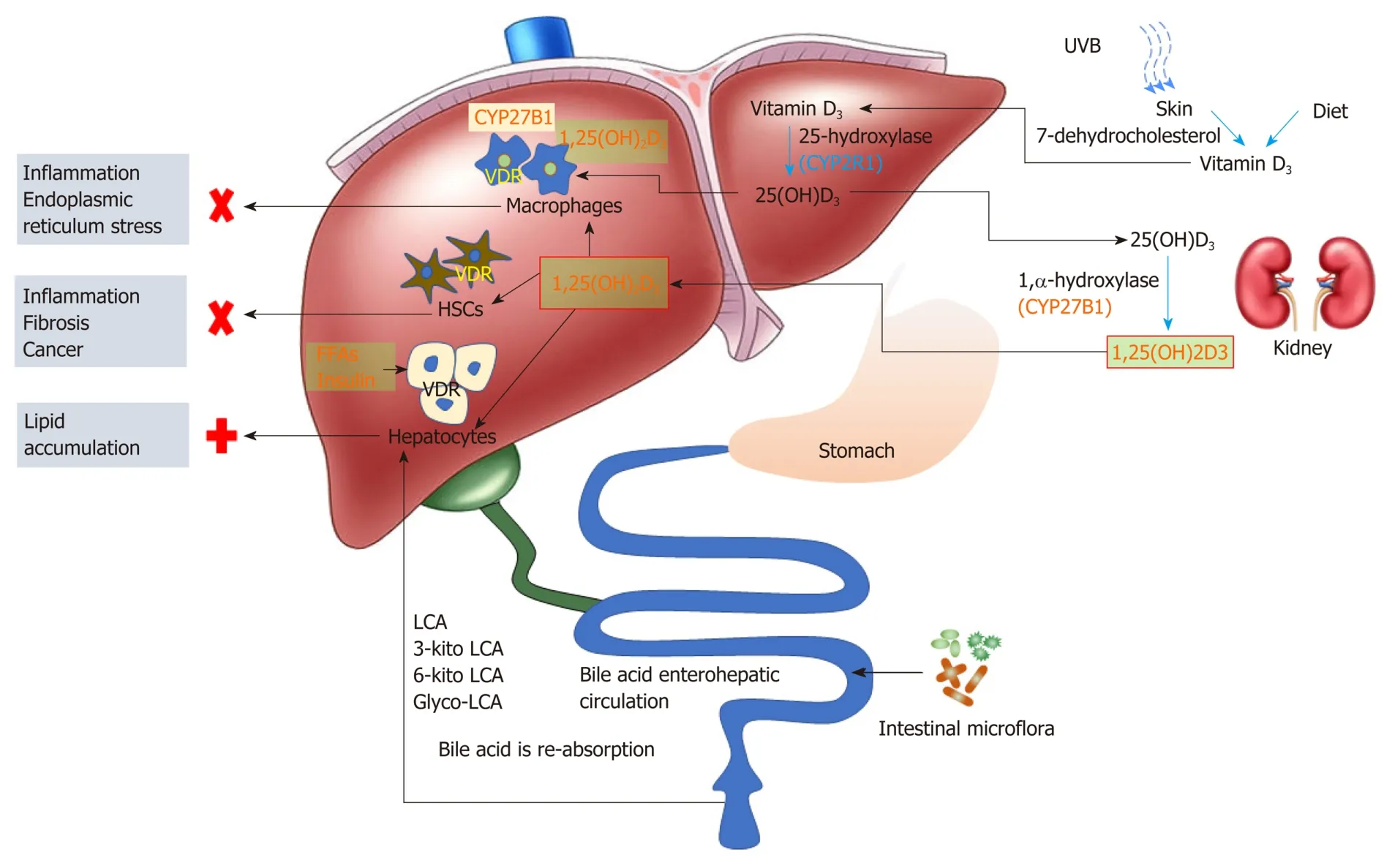
Figure 1 Expression and function of vitamin D receptor in different liver cells. Diet and skin producing pre-vitamin D (vitamin D3) is transferred to the liver, and converts into 25(OH)D3. Liver synthesized 25(OH)D3 is transported to the kidney, and converts into biochemically active 1,25(OH)2D3 via the catalase CYP27B1. 1,25(OH)2D3 then enters the circulation to distribute to tissues. Liver macrophages express CYP27B1, and vitamin D receptor (VDR) activation in liver macrophages can be achieved by both circulating and locally synthesized 1,25(OH)2D3; VDR in hepatocytes can be activated by circulating 1,25(OH)2D3 and gut microbiota-metabolized secondary bile acids. VDR activation in liver macrophages and hepatic stellate cells exerts anti-inflammatory and anti-fibrosis effect, respectively; whereas VDR activation in hepatocytes is supposed to contribute to lipid accumulation in the liver. LCA: Lithocholic acid; VDR: Vitamin D receptor; HSC: Hepatic stellate cells; UVB: Ultraviolet radiation B.
 World Journal of Gastroenterology2020年38期
World Journal of Gastroenterology2020年38期
- World Journal of Gastroenterology的其它文章
- Role of betaine in liver disease-worth revisiting or has the die been cast?
- Management of an endoscopy center during the outbreak of COVID-19: Experience from West China Hospital
- Gastrointestinal complications after kidney transplantation
- Acetyl-11-keto-β-boswellic acid inhibits proliferation and induces apoptosis of gastric cancer cells through the phosphatase and tensin homolog /Akt/ cyclooxygenase-2 signaling pathway
- Endogenous motion of liver correlates to the severity of portal hypertension
- Longitudinal decrease in platelet counts as a surrogate marker of liver fibrosis