
基于可逆动态共价化学的交联乙丙共聚物
2020-12-09 10:58:14刘姝慧
功能高分子学报 2020年6期
刘姝慧, 牛 慧
(大连理工大学高分子材料系,辽宁 大连 116024)
乙烯/丙烯无规共聚物(乙丙橡胶,EPR)作为一类密度小、耐热、耐老化、耐化学品腐蚀的合成橡胶材料,广泛应用在汽车工业、建筑材料、玩具等领域。随着现代社会的不断发展,EPR的消耗量迅猛增加,与此同时废旧EPR制品的回收问题也逐渐引起人们关注。上世纪80年代中期,全球橡胶制品回收率仅为1%,只有少数发达国家能对使用过的少量橡胶进行回收处理。近年来,随着人类环保意识的增强,美国、日本和欧盟等国家或组织已将橡胶的回收和循环再利用发展为实体产业,回收的废料不仅可以转化成新的能源或其他材料,还能带来可观的经济收益[1]。目前EPR制品回收方法仍以橡胶废料的后处理手段为主,例如通过热处理和机械处理相结合的方法来打破硫化交联形成的化学键,回收过程往往产生二次污染[2,3]。实现交联橡胶的绿色化回收再利用,尽可能避免处理过程引起的材料破坏,是科研人员和工业界共同追求的目标。
动态共价化学为制备具有可逆结构变化的聚合物提供了十分有效的方法。动态共价反应是分子之间通过共价键的形成-断裂而产生的可逆化学过程,在高分子合成中常采用的动态共价反应有Diels-Alder(D-A)反应,含二硫键、亚胺键、酰腙键的可逆反应和可逆自由基反应等,其在特定刺激(热、pH、光、氧化还原试剂等)条件下可在反应物与产物之间发生可逆的结构变化[4-7]。D-A反应作为一类有效的环加成反应,富电子的双烯体与缺电子的亲双烯体之间通过[4+2]环加成反应可形成稳定的六元环化合物,该反应无需催化剂,易实施。其中,呋喃/马来酰亚胺之间的D-A反应具有原料易得、加热易发生逆向D-A反应(rD-A)的特点,反应温和高效,成为目前构建可逆化学结构的研究热点[8-14]。 Picchioni课题组[15]于2015年首次报道了以马来酸酐接枝的EPR商业产品为原料,在聚合物侧基的酸酐上修饰呋喃基团将其转变为D-A反应物,从而使EPR具备可逆的交联功能,但受限于功能性基团含量低(摩尔分数不足1%),交联反应不易调控。迄今为止,以EPR为代表的饱和聚烯烃橡胶的D-A反应的报道一直较少,这主要是由于此类聚合物分子中仅含有饱和的脂肪族基团,可修饰性差,现有的少量报道多是基于含有功能性侧基的聚合物的后改性方法[9,16-18]。
最近,本课题组首次报道了通过Ziegler-Natta催化剂引发乙烯(E)、丙烯(P)和功能性单体8-呋喃-1-辛烯(FO)进行三元共聚,制得E/P/FO三元共聚物,这一方法可在较大范围内对E/P/FO中的呋喃含量(摩尔分数为0~9.7%)进行设计,并实现了E/P/FO的可逆交联[19,20]。本研究进一步将该共聚反应拓展至茂金属催化剂体系,考察FO参与的三元共聚反应规律,并以不同结构的双马来酰亚胺(BM)小分子为交联剂,研究了交联剂结构和浓度对E/P/FO热可逆交联效果和反复加工性能的影响。
1 实验部分
1.1 原料和试剂
四氢呋喃(THF):分析纯,天津市富宇精细化工有限公司,经溶剂精制系统处理后使用;呋喃:分析纯,上海麦克林生物化学有限公司,分子筛浸泡后使用;正丁基锂:上海阿拉丁生化科技股份有限公司;8-溴-1-辛烯:萨恩化学技术(上海)有限公司,分子筛浸泡后使用;无水乙醇、乙酸乙酯:分析纯,天津市富宇精细化工有限公司;无水硫酸钠:天津博迪化工股份有限公司;乙烯、丙烯:聚合级,大连光明特种气体有限公司;亚乙基桥二茚基二氯化锆(rac-Et(Ind)2ZrCl2):兰州石化公司提供,使用时配成4×10-3mol/L的甲苯溶液;甲基铝氧烷(MAO):安耐吉化学;甲苯:分析纯,天津市大茂试剂厂,氮气环境下用金属钠回流,二苯甲酮作指示剂,使用前蒸馏收集;十氢萘:分析纯,上海阿拉丁生化科技股份有限公司;N,N’-(亚甲基二苯基)双马来酰亚胺(Ph2):w=98%,萨恩化学技术(上海有限公司);1,6-双马来酰亚胺己烷(C6)和1,12-双马来酰亚胺十二烷(C12):参照文献[21]的步骤合成。
1.2 测试与表征
核磁共振氢谱(1H-NMR):FO单体测试(美国Bruker Avance Ⅱ 400型),以氘代氯仿、氘代二甲基亚砜为溶剂,测试温度25 °C;聚合物测试(美国Varian DLG400型),以氘代邻二氯苯为溶剂,测试温度100 °C。
差示扫描量热(DSC,美国TA公司Q2000型):在N2环境下,以10 °C/min的速率从室温逐渐升温至200 °C,在 200 °C恒温 5 min 消除热历史,再以 10 °C/min 的速率降温至-100 °C,最后以 10 °C/min 的速率升温至 200 °C。
凝胶渗透色谱(GPC,美国 Agilent公司 PL-GPC220型):流动相为 1,2,4-三氯苯,温度 150 °C,流速1.0 mL/min。
拉伸性能测试(美国Instron公司5567A型):夹具运行速率10 mm/min,室温。
1.3 FO的合成及表征
首先将呋喃的THF溶液温度降至-78 °C,逐滴加入正丁基锂溶液,升温至室温反应4 h后,再次降温至-78 °C,滴加8-溴-1-辛烯,置于室温下搅拌反应12 h。然后将反应产物倒入冰水中,用乙酸乙酯萃取,无水硫酸钠干燥。最后减压蒸馏除去杂质及副产物,得到产物 FO。1H-NMR (400 MHz, CDCl3) δ:7.29 (1H, d,C=CH), 6.28 (1H, t, C=CH), 5.97 (1H, d, C=CH), 5.82 (1H, m, C=CH), 4.98 (2H, m, C=CH2), 2.61 (2H, t,CH2), 2.05 (2H, m, CH2), 1.64 (2H, m, CH2), 1.35 (6H, m, CH2)。
1.4 E/P/FO三元共聚物的制备
E/P/FO的三元共聚反应在装有磁力搅拌的250 mL圆底烧瓶中进行。在干燥的烧瓶内通入E/P混合气体(0.1 MPa,nE/nP为1/1),依次加入溶剂甲苯 50 mL、FO单体(c=0.095 mol/L)和助催化剂MAO,搅拌 5 min后加入4 μmol茂金属催化剂rac-Et(Ind)2ZrCl2,在30 °C聚合反应15 min后将溶液倒入甲醇中,将析出的聚合物依次用乙醇和蒸馏水洗涤后,在50 °C的真空烘箱中干燥至恒重。聚合反应过程如图1所示。
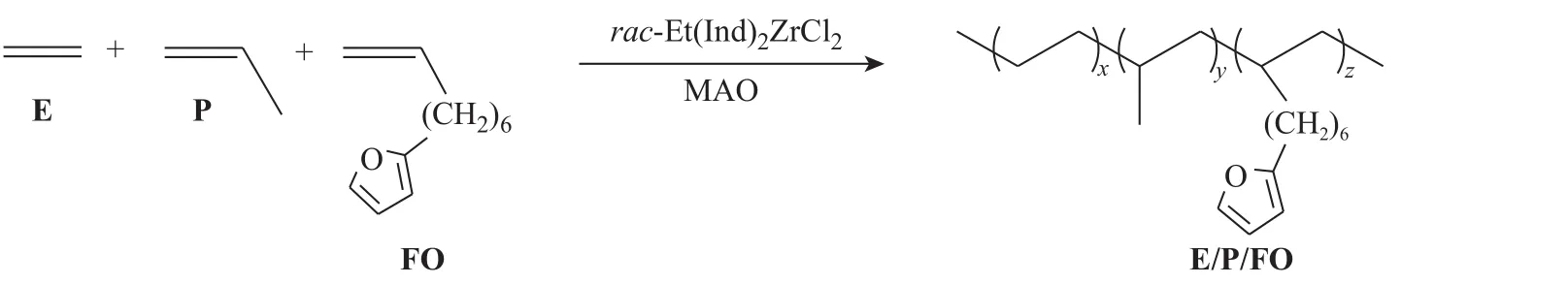
图1 E/P/FO三元共聚物的合成路线Fig. 1 Synthesis route of E/P/FO copolymer
1.5 交联聚合物的制备
将E/P/FO三元共聚物溶解在THF中,加入定量的小分子交联剂双马来酰亚胺,于80 °C反应4 h后,将溶剂干燥得到交联乙丙共聚物(CEP)。分别采用Ph2、C6和C12为交联剂,CEP的制备及重复加工过程如图2所示。将交联聚合物置于130 °C的热台上使聚合物解交联,用压机热压5 min成固定形状(压力10 MPa),随后将其置于80 °C的恒温箱中退火10 min使聚合物再次交联,得到用于测定凝胶含量和力学性能的试样。重复加工交联试样时,首先将试样剪碎成小块,然后将其置于130 °C的热台上进行热压,步骤同上。
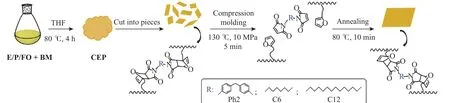
图2 CEP的制备及重复加工过程示意图Fig. 2 Schematic diagram of preparation and reprocessing of CEP
1.6 凝胶含量和溶胀比测试
将预先称好的试样(m1)浸泡在十氢萘中,并在室温下放置72 h至其达到溶胀平衡。随后取出溶胀样品,用纸巾吸干表面溶剂,立即称其质量(m2)。溶胀后的样品在80 °C下干燥24 h,得到恒定质量(m3)的样品。根据以下公式计算凝胶含量(G)和溶胀比(RS):

2 结果与讨论
2.1 E/P/FO的三元共聚
rac-Et(Ind)2ZrCl2/MAO催化下的E/P/FO三元共聚数据如表1所示。总体来看,共聚呈现出活性(每摩尔催化剂每小时生成的聚合物质量)高、FO转化率 (CFO)高的特点。呋喃基团中的O原子对茂金属催化剂的活性中心具有一定影响,但是当n(MAO)/n(FO) > 1时,E/P/FO三元共聚活性均高于E/P二元共聚活性,这可能是由于MAO中的Al原子与呋喃中的O原子之间存在Lewis酸-碱络合,使O原子无法对茂金属活性中心Zr产生毒害作用。
聚合物中 E、P、FO 的摩尔分数(χE,χP,χFO)通过1H-NMR 测定,E/P 二元共聚物(Run1)和典型的 E/P/FO三元共聚物(Run5)的1H-NMR谱图如图3所示。其中0.95~1.65处是主链中的乙烯、丙烯单元的CH3、CH2、CH特征峰,2.56处是与呋喃环相连的CH2特征峰,5.90、6.17、7.19处是呋喃环的特征峰,与二元共聚物的核磁谱图相比,三元共聚物的核磁谱图中,2.56处出现的特征峰表明FO成功插入到聚合物链中,合成了含有呋喃侧基的乙丙共聚物。通过特征基团的面积可以计算出共聚物组成,结果汇总在表1中。可以看出,nAl/nZr对共聚物组成特别是FO插入率无明显影响。在常规的MAO浓度范围内(nAl/nZr= 2 000~6 000),FO的转化率均可达60%以上。GPC测试结果(图4(a))表明三元共聚物数均分子量较二元共聚物有所下降。DSC测试结果(图4(b))表明,所得聚合物为无规共聚物,具有单一的玻璃化转变温度(Tg),位于-56~-61 °C 。
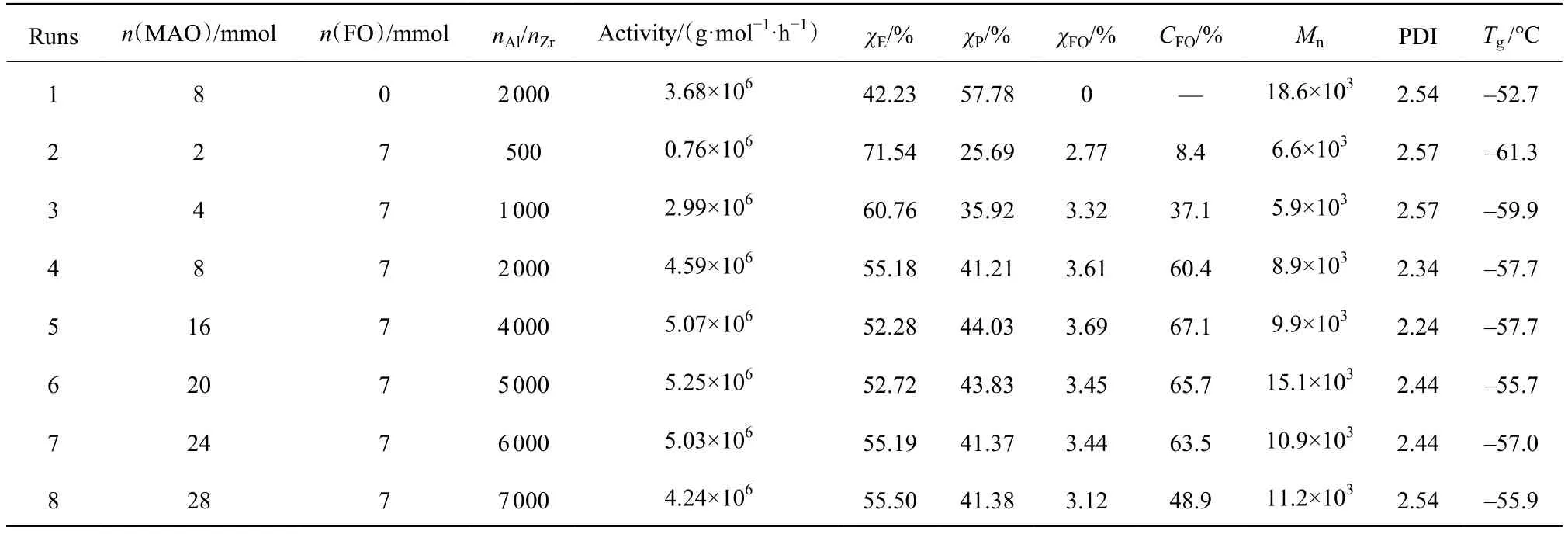
表1 E/P/FO三元共聚物的组成和结构Table 1 Compositions and structures of E/P/FO copolymer
2.2 可逆交联乙丙共聚物的制备及性能
选取Run5为原料,分别以3种不同结构的BM(C12、C6和Ph2)为交联剂,通过控制BM与呋喃基团的物质的量之比(nBM/nF),实施D-A反应得到一系列交联聚合物CEP,其凝胶含量和溶胀比列于表2中。由表2可知,交联剂浓度的增加可以显著提高交联网络的致密程度,随着nBM/nF从0.25/1增加至1/1,样品CEPC12凝胶含量从82.4%增至98.8%,溶胀比从655%降至142%,说明可以通过控制交联剂的用量来调控CEP的交联密度。同时,交联剂的结构对交联效果也有一定影响。从第一次加工样品的凝胶含量可以看出,采用尺寸较小的双马来酰亚胺Ph2和C6为交联剂得到的交联网络更为致密,其凝胶含量分别高达96.2%和98.2%,溶胀比也比C12为交联剂的样品更低。表2中还列出了上述样品经历3次加工成型后,凝胶含量和溶胀比的变化。结果表明,多次加工后CEP的交联程度仍恢复很好,这是由于D-A/rD-A可逆反应高效。此外,使用不同交联剂制备的样品,重复加工后其交联网络的稳定程度不尽相同。以柔性更好的C6和C12为交联剂所制备样品其交联程度更加稳定;而刚性较大的交联剂Ph2所得交联网络经历多次加工后,凝胶含量和溶胀比变化较大。
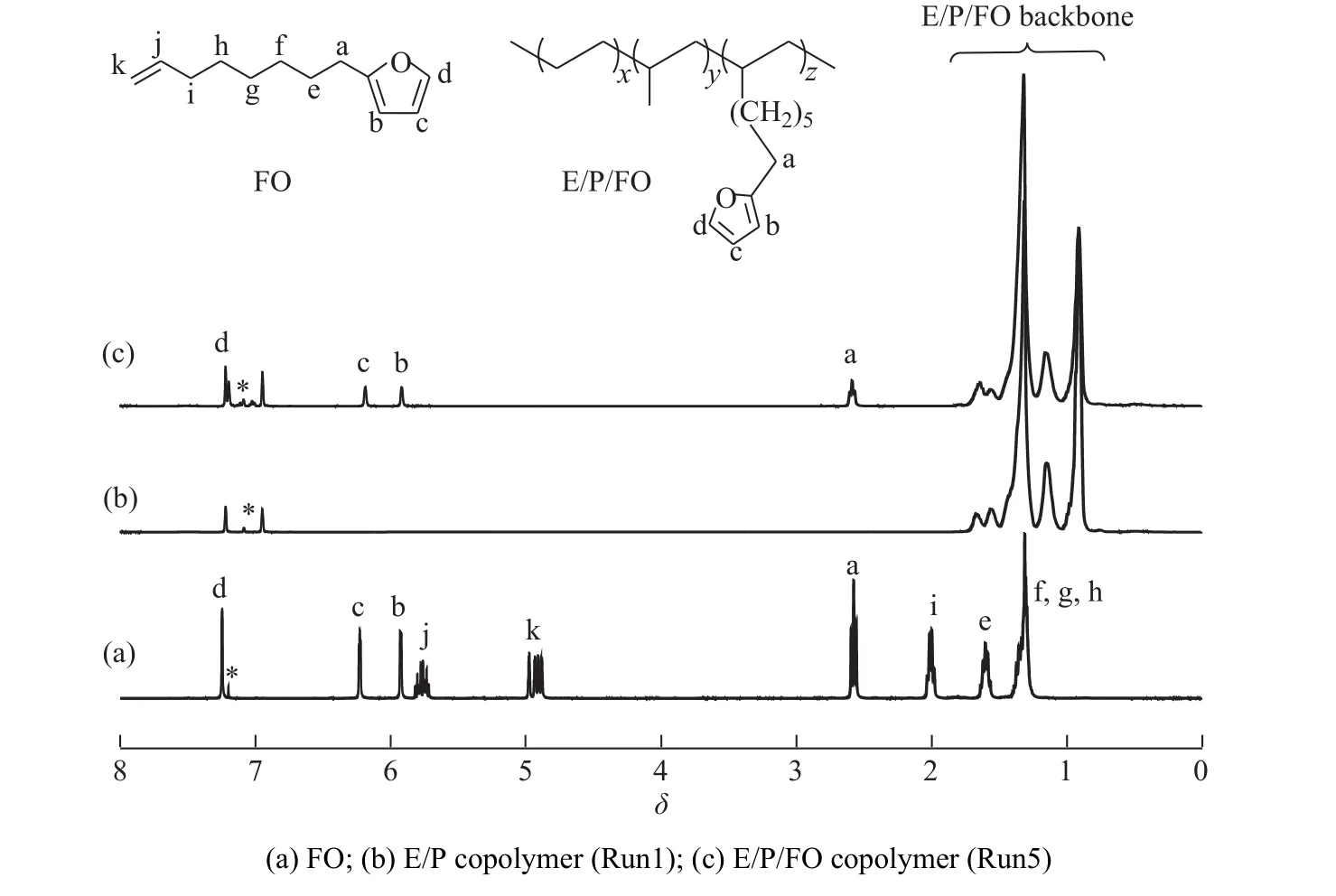
图3 单体和和聚合物的1H-NMR谱图Fig. 3 1H-NMR spectra of monomer and polymers
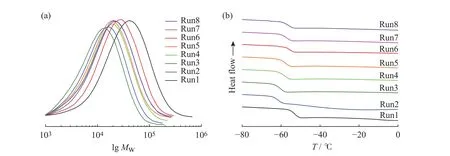
图4 聚合物的(a)GPC 曲线及(b)DSC 曲线Fig. 4 GPC curves (a) and DSC curves (b) of the polymers
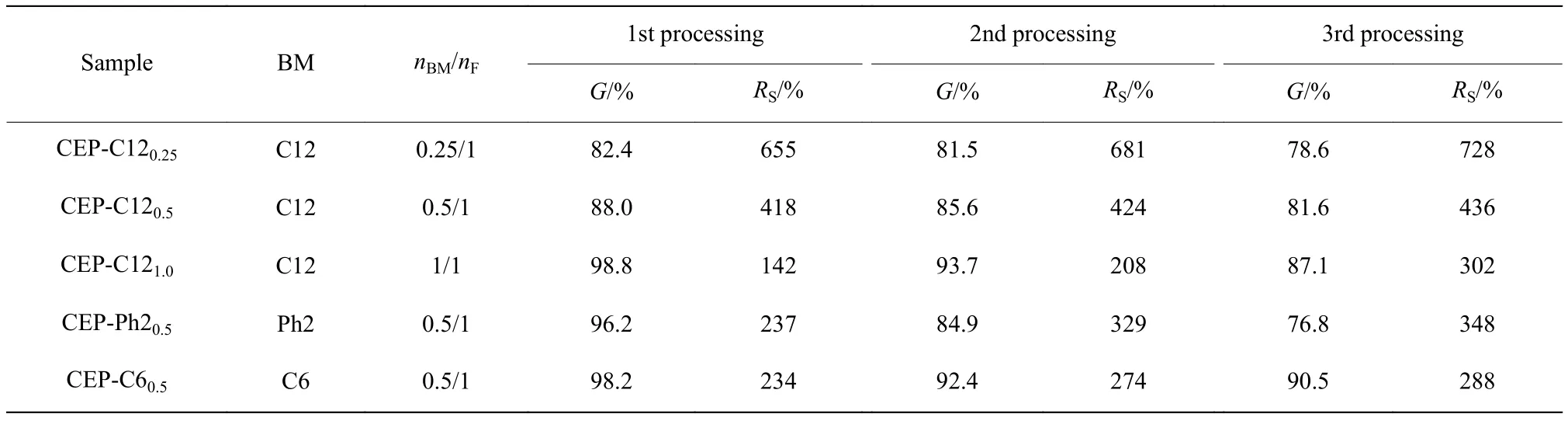
表2 CEP重复加工后的凝胶含量和溶胀比Table 2 Gel fraction and swelling ratio of CEP after repeated processing
对上述CEP样品进行拉伸性能测试,图5是CEP样品的应力-应变曲线,其中第1次加工所得样品(图5中的黑色实线)的拉伸性能数据如表3所示。对比CEP-C120.25、CEP-C120.5和CEP-C121.0的拉伸性能数据可以看出,随着交联剂用量增加,样品的交联密度增大,材料内部形成更加致密的三维网络,使材料断裂伸长率逐渐减小,断裂应力逐渐增大,杨氏模量增加。可见,通过调控交联剂的添加量可以设计材料结构,获得不同性能的交联产物。此外,使用不同结构交联剂制备的样品,其性能也有所差异。对比CEP-C120.5、CEP-Ph20.5和CEP-C60.5可以看出,以C6和Ph2为交联剂所制得样品的强度要明显高于以C12为交联剂制得的样品,这一规律与表2中溶胀比数据的规律一致,本文认为这是由于体积较小的交联剂所形成的交联网络更致密,从而赋予材料更高的强度。
将上述进行拉伸性能测试后的样品剪碎成小块,放置于130 °C的热台上,样品中的呋喃-马来酰亚胺D-A加成产物受热后发生rD-A反应引起材料解交联,从而使其能够再次热压成型;样品冷却后交联结构可再次生成。对所得样品进行第2次拉伸实验,其应力-应变曲线如图5中蓝色虚线所示。重复上述步骤对材料进行第3次加工,其应力-应变曲线如图5中绿色点线所示。可以看出,样品CEP-C120.25和CEP-C120.5均表现出优异的重复加工性能,材料的力学性能重现性良好,而样品CEP-C121.0的回复性能较差。这是由于CEP-C121.0中的马来酰亚胺和呋喃基团数量相同,对于小分子交联剂而言,当其一端完成D-A反应后,另一端运动能力受限,而其周围恰好存在未反应的呋喃基团的几率不高,造成样品交联程度不稳定;样品CEP-C120.25和CEP-C120.5中,聚合物上的呋喃基团相对于交联剂中的马来酰亚胺基团是过量的,有利于交联剂充分反应,保证了反复加工后样品交联程度的稳定。同时,柔性的交联剂分子能促进交联反应高效进行,这从CEP-C60.5优异的重复加工效果也能得到验证;与此相反,在样品CEP-Ph20.5中,具有刚性结构的交联剂则不利于材料反复加工中的性能稳定。
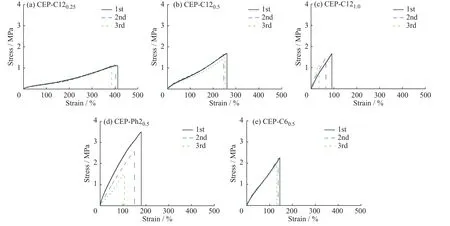
图5 CEP重复加工后的应力-应变曲线Fig. 5 Stress-strain curves of CEP after repeated processing
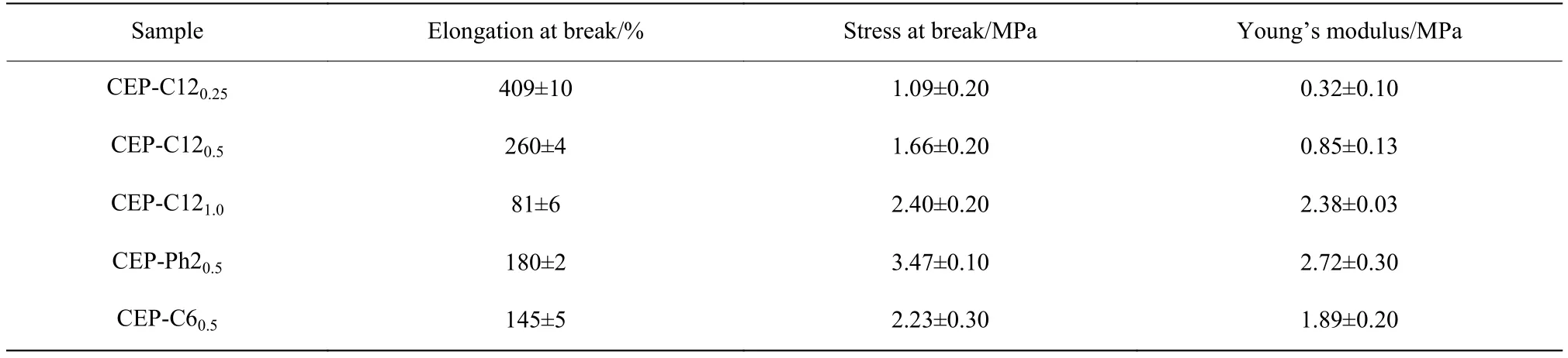
表3 CEP的拉伸性能Table 3 Tensile properties of CEP
3 结 论
(1)利用茂金属催化体系rac-Et(Ind)2ZrCl2/MAO引发E/P/FO的三元共聚,成功合成了侧基含呋喃取代基的EPR,通过调控MAO与FO的物质的量之比,可使共聚反应呈现活性高、FO转化率高的特点。
(2)将含呋喃取代基的EPR分别与3种双马来酰亚胺小分子交联剂(C12、C6和Ph2)进行Diels-Alder反应制备了一系列可逆交联聚合物,通过改变交联剂结构及马来酰亚胺与呋喃的物质的量之比,可调节聚合物的交联程度。
(3)马来酰亚胺和呋喃基团之间Diels-Alder反应的热可逆性赋予了交联聚合物可进行多次重复加工的功能,采用具有柔性结构的交联剂C12和C6可获得性能更稳定的可逆交联乙丙共聚物。
猜你喜欢
民用飞机设计与研究(2020年1期)2020-05-21 07:24:52
小学生学习指导(低年级)(2017年11期)2017-10-23 01:48:55
中学生(2017年13期)2017-06-15 12:57:49
中国塑料(2017年2期)2017-05-17 06:13:21
烟草科技(2015年8期)2015-12-20 08:27:14
中国塑料(2015年6期)2015-11-13 03:02:49
传奇故事(破茧成蝶)(2015年1期)2015-02-28 09:26:59
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
无机化学学报(2014年12期)2014-02-28 17:34:06
无机化学学报(2014年6期)2014-02-28 17:32:06
