
树突状细胞在心肌梗死及其修复重构中的作用*
2020-12-03 14:22:40张右铭刘海波
中国病理生理杂志 2020年11期
张右铭, 刘海波,2△
树突状细胞在心肌梗死及其修复重构中的作用*
张右铭1, 刘海波1,2△
(1同济大学附属东方医院心内科,上海 200120;2复旦大学附属中山医院青浦分院心内科,上海 201700)
树突状细胞;心肌梗死;动脉粥样硬化;缺血再灌注损伤;心肌重构
虽然随着介入技术的发展,及时的再灌注治疗降低了急性心肌梗死的死亡率,但心梗后心脏的缺血再灌注损伤及不良重构,导致患者发生慢性心力衰竭,生活质量急剧下降,对社会经济造成了严重的负担。所以如何预防心肌梗死和改善心梗后缺血损伤及不良重构引起的心力衰竭,提高患者预后及生存质量成了新的热门研究话题。
目前的研究发现,炎症与心肌梗死(myocardial infarction, MI)的发生发展关系密切,冠心病发生的根本原因是动脉粥样硬化,目前研究表明炎症参与了动脉粥样硬化的整个进程[1],随着慢性炎症和脂质斑块的累积,不断缓慢进展,斑块破裂或侵蚀导致血栓形成,阻塞血管并导致MI的发生,MI后,坏死凋亡的心肌细胞可激活损伤相关分子模式(damage-associated molecular patterns, DAMPs),通过释放各种细胞因子及趋化因子,招募和激活各种免疫细胞,参与MI后心肌损伤的修复[2]。
树突状细胞(dendritic cells, DCs)作为重要的抗原呈递细胞,能调节固有免疫和适应性免疫的各种炎症细胞,从而在免疫反应中发挥重要的作用[3]。根据表面标志物的表达,DCs可以被粗略地分为经典树突状细胞(conventional dendritic cells, cDCs)及浆细胞样树突状细胞(plasmacytoid dendritic cells, pDCs)两大类,可用不同的表面标志物来区分,其中cDCs还可细分为cDCs1和cDCs2两个亚型[4-6],详见表1。不同的实验使用了不同的方法来特异性清除小鼠体内的DCs或其亚型。近来,越来越多的实验发现DCs在动脉粥样硬化和MI后炎症反应中发挥重要作用。因此,本文就DCs在MI全过程中各个阶段的作用进行综述,重点阐述DCs在动脉粥样硬化、MI后心肌缺血再灌注损伤(myocardial ischemia-reperfusion injury, MI/RI)和心室重构中的作用。
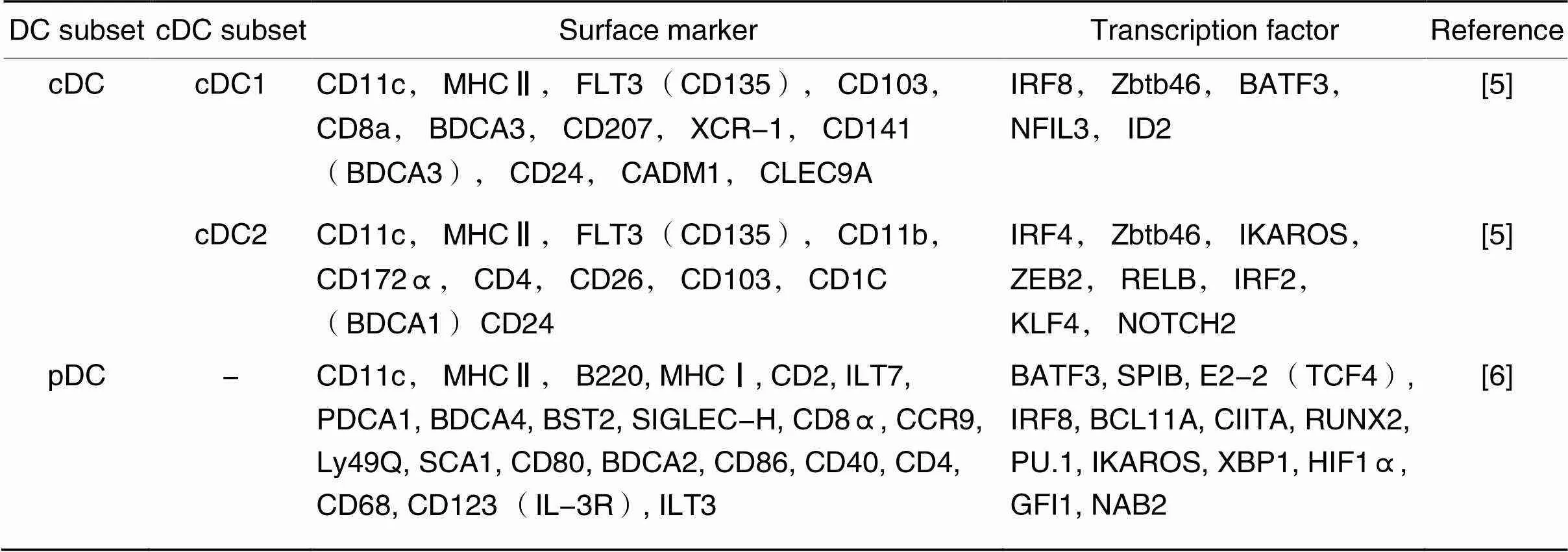
表1 DCs分类及常见表面标志物
1 DCs在动脉粥样硬化中的作用
动脉粥样硬化是一个复杂的终生过程,涉及先天和后天免疫反应及慢性炎症,是引起缺血性心脏病和脑梗死最常见的原因[7]。动脉粥样硬化斑块的缓慢形成是无症状的,其特征是内皮功能障碍以及低密度脂蛋白(low-density lipoprotein, LDL)、免疫细胞和坏死碎片在内皮下间隙的积聚。内皮细胞激活触发白细胞黏附分子的表达,后者通过CCR2发出信号,刺激炎症性单核细胞的迁移和浸润[8]。沉积在内膜的LDL可被进一步氧化修饰为氧化低密度脂蛋白(oxidized low-density lipoprotein, oxLDL),发挥抗原作用,被吞噬形成泡沫细胞后,可引发动脉粥样硬化的形成,在这一过程中,巨噬细胞占主导作用[9]。虽然巨噬细胞在动脉粥样硬化的细胞浸润中占主导地位,但研究人员在小鼠动脉粥样硬化模型的主动脉内膜[10]和斑块内[11],均发现了CD11c+MHCII+DCs的聚集。并且,在人类的动脉内膜中[12]和斑块内[13]也可找到大量CD1α+、CD83+及CD86+DCs,其主要存在于斑块肩部以及斑块核心的边缘部位[13-14]。同样有研究表明,在人为制造高胆固醇血症几天后,低密度脂蛋白受体基因敲除(low-density lipoprotein receptor gene knockout,-/-)小鼠的内膜血管CD11c+DCs内就可以发现脂质的积聚,并呈现泡沫细胞样外观,参与早期阶段斑块的形成[15]。还有研究发现,无论在-/-小鼠模型或载脂蛋白E基因敲除(apolipoprotein E gene knockout,-/-)小鼠模型中,耗竭CD11c+DCs都会导致血浆总胆固醇水平的升高。人为延长-/-或-/-小鼠模型中DCs寿命和免疫原性后,血浆LDL和极低密度脂蛋白(very-low-density lipoprotein, VLDL)水平明显降低,但与对照组相比,小鼠动脉粥样硬化斑块的大小并没有明显改变[16]。这一过程的具体分子机制尚不清楚,不过可以看出DCs同样具有摄取脂质的能力并参与泡沫细胞的形成。
此外,也有研究发现,人X组分泌磷脂酶A2修饰的低密度脂蛋白(low-density lipoprotein modified by human group X-secreted phospholipase A2, LDLx)和oxLDL可通过DCs激活T细胞[17],提呈抗原并启动特异性免疫被认为是DCs在动脉粥样硬化中的重要功能。先前研究发现,分离的主动脉CD11c+DCs暴露于血液中的抗原后,可在体外诱导抗原特异性T细胞增殖[18-19],并且主动脉CD11c+DCs可在体外促进抗原T细胞产生肿瘤坏死因子α (tumor necrosis factor-α,TNF-α)和干扰素γ (interferon-γ, IFN-γ)[20],提示DCs可引起局部T细胞的活化和促炎细胞因子的产生。
不过,DCs对动脉粥样硬化起抑制还是促进的作用却存在争议。有实验报道人为删除-/-小鼠中的CD74 (一种参与MHCⅡ复合物形成的关键蛋白),间接减少DCs的数量后,发现T细胞的激活及斑块的形成减少了[21]。然而,另一实验发现在人为制造的-/--/-小鼠中,起保护起作用的调节性T细胞(regulatory T cells, Treg)减少,促动脉粥样硬化的CD8+T细胞增加,最终促进了动脉粥样硬化斑块的形成[22]。
同样的矛盾结果还体现在对DCs亚型功能的研究中。有研究将含锌指及BTB结构域蛋白46 (zinc finger and BTB domain containing 46,)-白喉毒素受体(diphthera toxin receptor,)转基因小鼠的骨髓移植到-/-小鼠模型中,注射白喉毒素选择性清除-/-小鼠体内cDCs,发现斑块大小不受影响[23]。但在利用FMS样酪氨酸激酶3 (FMS-like tyrosine kinase, Flt3)制造-/--/-小鼠模型、特异性耗竭CD103+cDCs的实验中,发现小鼠体内Treg减少,加重了动脉粥样硬化,提示cDCs对动脉粥样硬化有抑制作用[24]。同样,在利用碱性亮氨酸拉链ATF样转录因子3 (basic leucine zipper ATF-like transcription factor 3, Batf3)制造-/--/-小鼠模型、特异性耗竭cDCs的实验中,斑块大小没有明显变化[25]。但在基于-/--/-小鼠模型的实验中却发现cDCs可以通过刺激Th1细胞来促进动脉粥样硬化斑块形成[26]。在针对pDCs的实验中,使用大量抗骨髓基质细胞抗原2 (bone marrow stromal antigen 2, BST2)的抗体来清除pDCs,发现清除pDCs促进了-/-小鼠的动脉粥样硬化[27],但减轻了-/-小鼠的动脉粥样硬化[28]。同样,有研究将血树突状细胞抗原2 (blood dendritic cell antigen 2,)-转基因小鼠的骨髓移植到-/-小鼠模型中,注射白喉毒素选择性清除-/-小鼠体内pDCs,发现清除pDCs促进了斑块发展[29],但在-/--小鼠模型中,斑块大小没有变化[30]。
造成这些差异的原因目前尚不清楚,不过由于-/-和-/-小鼠模型中脂质分布的不同和对免疫细胞效应的不同,两者有各自的优缺点[31],所以造成这些相互矛盾结果的原因可能是由于小鼠模型的不同。此外,于永慧等[32]发现,不同阶段-/-小鼠的炎症基因存在差异化表达且不同周龄-/-小鼠中炎症因子表达情况有显著差异。因此不同实验中小鼠模型周龄的不同也可能是造成不同结果的重要因素。另外,考虑到用来标记DCs及其亚型的分子(如MHCⅡ、CD103、CD8a等)也可以被其他免疫细胞表达,因此不能排除受到了其他免疫细胞的影响。并且由于模型的限制,无法维持长期的特异性耗竭,也无法有效评价DCs及其亚型在慢性炎症中的作用,更有效的特异性耗竭DCs及其亚型的办法仍需进一步研究。
2 DCs在MI/RI中的作用
随着介入技术的发展,MI患者的生存率大大提高,然而,缺血心肌的血流恢复过程亦会引起损伤,这种现象被称为MI/RI,它会降低心肌再灌注的有益影响[33]。MI/RI的发病机制很复杂,炎症反应在这一过程中扮演了重要角色。除了中性粒细胞、巨噬细胞和淋巴细胞外, DCs在缺血再灌注损伤中也发挥了重要作用。已有文献报道,人DCs中P2Y11受体具有免疫抑制作用,P2Y11受体激动剂可以减轻缺血再灌注过程中的炎症反应并保护缺血器官[34]。但也有研究发现,在大鼠心肌缺血再灌注过程中,CD1a+CD80+DCs向心肌的迁移、黏附和聚集增加,加重了心肌损伤[35]。
不过,目前针对DCs在MI/RI中作用的研究相对较少,尚未能有确切的结论。但在小鼠研究中有一致的证据表明,促炎CD4+T细胞的浸润加重了MI/RI。除了CD4+T细胞外, CD8+记忆T细胞亚群在再灌注开始后早期就聚集在冠状动脉微循环中,可能参与到再灌注过程中,对心肌产生损害[36-37]。考虑到DCs作为强大的抗原提呈细胞,是特异性免疫应答的始动者,所以在MI/RI过程中,各类T细胞的浸润很大可能是通过DCs发动。已有证据表明,在肝脏、肾脏等器官移植时发生的缺血再灌注损伤中, DCs可以激活T细胞,加强炎症对再灌注后组织的损伤[38-39]。
亦有实验发现,在结扎小鼠冠状动脉后,坏死心肌和正常心肌之间的梗死边界区内CD11c+DCs数量明显增加,并且和CD4+T细胞形成集落[40]。这些实验均提示在MI时, DCs与T细胞有某种联系,但其确凿的实验证据和内在的分子机制仍有待进一步研究。
3 DCs在心室重构中的作用
MI后左室重构是一个复杂的心肌结构改变过程,不良左室重构严重影响患者的预后。已有大量研究表明,炎症在心肌梗死愈合和随后的左室重构中起着至关重要的作用[41-42]。
Anzai等[43]的研究设计了条件性DCs敲除小鼠,将人DTR结合在DCs的启动子区域,在小鼠MI术前采用注射白喉毒素的方法完全清除小鼠体内的CD11c+DCs,发现在DCs敲除小鼠MI模型中,其左室重构较对照组明显恶化,虽然病理检查未显示梗死面积有明显区别,但DCs敲除小鼠梗死区域心脏壁更薄,并且新生血管受损。他们更深入的研究发现,梗死区及周边区域浸润高表达Ly6C (Ly6Chigh)的单核细胞和M1型巨噬细胞明显增加,而低表达Ly6C (Ly6Clow)的单核细胞和M2型巨噬细胞显著降低,并且在DCs敲除MI模型小鼠中,炎症因子和MMP-9的表达也明显增高,而抗炎因子IL-10却显著降低。这些研究结果表明DCs敲除后通过激活炎症单核细胞和M1型巨噬细胞及抑制抗炎单核细胞和M2型巨噬细胞,增强了炎症反应及细胞外基质的降解,从而延缓MI后的心脏愈合,导致心功能恶化。这些研究提示DCs可通过调节单核细胞和巨噬细胞的动态平衡及转化而起到抑制心脏重构的作用。
有研究对ST段抬高心肌梗死死亡患者进行尸检,发现心脏破裂组与非破裂组相比, CD68+巨噬细胞浸润增加, CD209+DCs和CD11c+DCs浸润及修复性纤维化程度较低,提示心梗部位DCs数量的减少与巨噬细胞浸润增加会使修复性纤维化受损,增加心肌梗死后心脏破裂的风险,表明DCs在心肌梗死后炎症及随后的愈合过程中具有保护作用[44]。我们前期的研究发现,MI小鼠模型中CD11c+DCs产生的外泌体可将CD4+辅助性T细胞招募到心肌梗死区,并有助于预防心肌梗死后的左心室不良重构[45]。这些结果均提示DCs在MI后炎症反应和随后的左心室重构中具有心肌保护作用,但具体针对DCs各亚型的功能,不同实验报告了不同的结果。
有研究利用-重组小鼠,注射白喉毒素选择性地清除CD103+及CD11b+cDCs,发现与对照组相比,选择性清除cDCs显著减少了MI后心肌损伤的范围,且能预防心室不良重构,改善心功能,表明cDCs不利于MI后心肌损伤的恢复及心功能的改善,加重了心室不良重构[46]。但也有实验发现,心肌梗死后, XCR-1+CD172α+cDCs浸润心肌,摄取坏死心肌细胞释放的α-肌球蛋白碎片,逐渐迁移到纵膈淋巴结中并将心脏自身抗原提呈给CD4+T细胞,并最终引起Treg的增殖[47]。活化的Treg可通过增加巨噬细胞精氨酸酶1、IL-13、骨桥蛋白及TGF-β的表达而诱导巨噬细胞向M2型分化,从而抑制了MI后的心脏重构[48];而耗竭Treg可加速心肌梗死后心室的扩张,加重心室重构[49]。由此可以推测cDCs对心室功能有保护作用。
为了探讨pDCs的作用,研究者在-重组小鼠中注射白喉毒素选择性地耗竭pDCs,发现对MI后心功能没有影响,其作用可能在于产生I型干扰素并保护组织免受病毒感染,在MI后心脏修复的过程中并不重要[46]。
整体来说,DCs在预防心室不良重构方面发挥了积极的作用,但在针对DCs不同亚群的研究中,不同实验却得出了矛盾的结论,有待进一步研究。
4 基于DCs的免疫疗法
近年来,人们越来越意识到炎症在整个心梗过程中的重要性,研究人员一直以来都尝试开发基于免疫炎症的新型疗法,并且已收获很多成果[50-52]。DCs作为免疫调控的中心环节,深入参与了MI发生发展的全过程,因此基于DCs的免疫疗法潜力巨大。
将携带人载脂蛋白B100 (apolipoprotein B100, ApoB100)的致耐受树突状细胞(tolerogenic dendritic cells, tDCs)导入-/-小鼠中,使之成为具有人基因的转基因-/-小鼠(tg×-/-小鼠),可显著降低小鼠体内效应T细胞的数量并诱导Treg的增殖,从而显著延缓动脉粥样硬化病变的发展[53]。这提示基于DCs的免疫治疗可以延缓动脉粥样硬化的发展,在预防MI方面有极大潜力。同样,李大主等[54]将携带热休克蛋白60 (heat shock protein 60, HSP60)的tDCs接种至-/-小鼠后,发现小鼠动脉粥样硬化斑块中的炎症反应和斑块的进展受到抑制。后续有研究者发现,经典调脂药物——他汀类药物可以通过抑制人CD83+CD86+DCs中的miRNA let-7c来抑制oxLDL诱导DCs的成熟,进而抑制DCs促进T细胞增殖的能力,从而影响动脉粥样硬化斑块的发生和发展[55]。这揭示了他汀类药物治疗动脉粥样硬化时还具有免疫治疗方面的作用。最近,研究者基于CX3CL1/CX3CR1通路开发了一种针对CX3CR1的DCs靶向DNA疫苗,该疫苗通过将质粒与针对DCs的限制性抗原摄取受体DEC205的单链Fv抗体(scFv)结合,从而将质粒特异性提供给DCs,增强接种效果,并用于小鼠动脉粥样硬化模型。研究发现,注射修饰后的DEC205-CX3CR1 DNA疫苗显著减少了小鼠斑块处巨噬细胞的浸润,减小了小鼠粥样硬化斑块的大小,对动脉粥样硬化具有显著的抑制作用[56]。研究者已建议使用DNA疫苗阻断CX3CR1通路的方法作为目前动脉粥样硬化治疗方法的补充。
有研究者发现,刺激人DCs内的P2Y11受体对MI/RI具有保护作用,在器官移植和急性MI后的MI/RI期间, P2Y11受体激动剂药物可以提供有益帮助[34]。此外,有研究者发现,高迁移率族蛋白B1(high mobility group protein B1, HMGB1)作为MI/RI后心肌损伤产生的DAMPs,通过Toll样受体4 (Toll-like receptor 4, TLR4)通路激活CD1a+CD80+DCs,影响其在心肌中的分布,诱导DCs活化和成熟。并且,使用HMGB1中和抗体可提供明显的心脏保护[35]。所以说,基于DCs的免疫疗法在减轻MI/RI方面亦有重要作用。
早期有研究通过敲除小鼠白细胞介素1受体相关激酶4 (interleukin-1 receptor-associated kinase 4,-)基因,消除CD11c+DCs动员、捕获抗原、成熟及产生细胞因子的能力,从而使--/-MI模型小鼠心功能及心室不良重构获得改善,存活率提高[57]。使用TNF-α和心脏抗原刺激骨髓源性树突状细胞(bone marrow-derived dendritic cells, BMDCs)以制备tDC,再向MI小鼠模型中注入tDC,可激活小鼠体内的Treg,进而促进早期巨噬细胞亚群从炎性M1型转换为修复性M2型,并增加新生血管的生成,从而减少梗死面积,提升左室功能[52]。除此之外,我们前期的研究还发现,通过体外给予MI模型小鼠CD11c+DCs分泌的外泌体治疗,可以激活CD4+T淋巴细胞并改善心脏功能[45]。同样,经典药物血管紧张素转化酶抑制剂对CD11c+DCs介导的免疫炎症反应也有抑制作用,可以减轻MI后的炎症反应,从而改善心室功能,提高生存率[58]。
各种实验结果均表明,基于DCs的免疫疗法有十分光明的前景,相信随着后续研究的不断探索,免疫治疗将在MI的预防与治疗中发挥重要作用。
5 总结与展望
DCs在心肌梗死的发生和发展的各个阶段都发挥着重要的作用,但现阶段的研究对其在各个阶段中的具体作用及相应的分子机制,尚不能达成共识,仍需进一步研究。基于DCs的免疫疗法前景广阔,目前已取得一定成果,这为延缓动脉粥样硬化的发展、减轻急性心肌梗死后缺血再灌注损伤和预防心脏不良重构提供了全新的思路,具有巨大临床应用潜力。
[1] Gistera A, Hansson GK. The immunology of atherosclerosis[J]. Nat Rev Nephrol, 2017, 13(6):368-380.
[2] Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling [J]. Nat Rev Cardiol, 2014, 11(5):255-265.
[3] Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease[J]. Nat Rev Immunol, 2017, 17(1):30-48.
[4] Guilliams M, van de Laar L. A Hitchhiker's guide to myeloid cell subsets: practical implementation of a novel mononuclear phagocyte classification system[J]. Front Immunol, 2015, 6:406.
[5] Sichien D, Lambrecht BN, Guilliams M, et al. Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues[J]. Mucosal Immunol, 2017, 10(4):831-844.
[6] Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells[J]. Nat Rev Immunol, 2015, 15(8):471-485.
[7] Herrero-Fernandez B, Gomez-Bris R,Somovilla-Crespo B, et al. Immunobiology of atherosclerosis: a complex net of interactions[J]. Int J Mol Sci, 2019, 20(21):5293.
[8] Gil-Pulido J, Zernecke A. Antigen-presenting dendritic cells in atherosclerosis [J]. Eur J Pharmacol, 2017, 816:25-31.
[9] Back M, Yurdagul A, Jr., Tabas I, et al. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities [J]. Nat Rev Cardiol, 2019, 16(7):389-406.
[10] Busch M, Westhofen TC, Koch M, et al. Dendritic cell subset distributions in the aorta in healthy and atherosclerotic mice[J]. PLoS One, 2014, 9(2):e88452.
[11] Sage AP, Murphy D, Maffia P, et al. MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity[J]. Circulation, 2014, 130(16):1363-1373.
[12] Millonig G, Niederegger H, Rabl W, et al.Network of vascular-associated dendritic cells in intima of healthy young individuals[J]. Arterioscler Thromb Vasc Biol, 2001, 21(4):503-508.
[13] Yilmaz A, Lochno M, Traeg F, et al. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques[J]. Atherosclerosis, 2004, 176(1):101-110.
[14] Erbel C, Sato K, Meyer FB, et al.Functional profile of activated dendritic cells in unstable atherosclerotic plaque[J]. Basic Res Cardiol, 2007, 102(2):123-132.
[15] Paulson KE, Zhu SN, Chen M, et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis[J]. Circ Res, 2010, 106(2):383-390.
[16] Gautier EL, Huby T, Saint-Charles F, et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis[J]. Circulation, 2009, 119(17):2367-2375.
[17] Liu A, Ming JY, Fiskesund R, et al. Induction of dendritic cell-mediated T-cell activation by modified but not native low-density lipoprotein in humans and inhibition by annexin a5: involvement of heat shock proteins[J]. Arterioscler Thromb Vasc Biol, 2015, 35(1):197-205.
[18] Weber C, Meiler S, Doring Y, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice [J]. J Clin Invest, 2011, 121(7):2898-2910.
[19] Choi JH, Do Y, Cheong C, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves[J]. J Exp Med, 2009, 206(3):497-505.
[20] Koltsova EK, Garcia Z, Chodaczek G, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis[J]. J Clin Invest, 2012, 122(9):3114-3126.
[21] Sun J, Hartvigsen K, Chou MY, et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice[J]. Circulation, 2010, 122(8):808-820.
[22] Wigren M, Rattik S, Yao Mattisson I, et al. Lack of ability to present antigens on major histocompatibility complex class II molecules aggravates atherosclerosis in-/-mice[J]. Circulation, 2019, 139(22):2554-2566.
[23] Rombouts M, Cools N, Grootaert MO, et al. Long-term depletion of conventional dendritic cells cannot be maintained in an atherosclerotic Zbtb46-DTR mouse model[J]. PLoS One, 2017, 12(1):e0169608.
[24] Choi JH, Cheong C, Dandamudi DB, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis[J]. Immunity, 2011, 35(5):819-831.
[25] Gil-Pulido J, Cochain C, Lippert MA, et al. Deletion of Batf3-dependent antigen-presenting cells does not affect atherosclerotic lesion formation in mice[J]. PLoS One, 2017, 12(8):e0181947.
[26] Li Y, Liu X, Duan W, et al. Batf3-dependent CD8α+dendritic cells aggravates atherosclerosis via Th1 cell induction and enhanced CCL5 expression in plaque macrophages[J]. EBioMedicine, 2017, 18:188-198.
[27] Daissormont IT, Christ A, Temmerman L, et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity[J]. Circ Res, 2011, 109(12):1387-1395.
[28] Macritchie N, Grassia G, Sabir SR, et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice [J]. Arterioscler Thromb Vasc Biol, 2012, 32(11):2569-2579.
[29] Yun TJ, Lee JS, Machmach K, et al. Indoleamine 2,3-dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T cells[J]. Cell Metab, 2016, 23(5):852-866.
[30] Mandl M, Drechsler M, Jansen Y, et al. Evaluation of the BDCA2-DTR transgenic mouse model in chronic and acute inflammation[J]. PLoS One, 2015, 10(8):e0134176.
[31] Getz GS, Reardon CA. Do the-/-and-/-mice yield the same insight on atherogenesis?[J]. Arterioscler Thromb Vasc Biol, 2016, 36(9):1734-1741.
[32] 于永慧,董瑞红,刘剑刚,等. 不同阶段-/-小鼠动脉粥样硬化炎症差异基因表达的比较研究[J]. 中国病理生理杂志, 2019, 35(9):1694-1699.
Yu YH, Dong RH, Liu JG, et al. Preliminary comparison of inflammatory differential gene expression during atherosclerosis in-/-mice[J]. Chin J Pathophysiol, 2019, 35(9):1694-1699.
[33] Yellon DM, Hausenloy DJ. Myocardial reperfusion injury[J]. N Engl J Med, 2007, 357(11):1121-1135.
[34] Chadet S, Ivanes F, Benoist L, et al. Hypoxia/reoxygenation inhibits P2Y11 receptor expression and its immunosuppressive activity in human dendritic cells[J]. J Immunol, 2015, 195(2):651-660.
[35] Xue J, Ge H, Lin Z, et al. The role of dendritic cells regulated by HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion injury[J]. J Cell Mol Med, 2019, 23(4):2849-2862.
[36] Boag SE, Das R, Shmeleva EV, et al. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients[J]. J Clin Invest, 2015, 125(8):3063-3076.
[37] Hoffmann J, Shmeleva EV, Boag SE, et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients[J]. Circ Res, 2015, 116(1):87-98.
[38] Funken D, Ishikawa-Ankerhold H, Uhl B, et al.targeting of dendritic cells sets tolerogenic environment and ameliorates CD4+T-cell response in the postischemic liver[J]. FASEB J, 2017, 31(11):4796-4808.
[39] Ozaki KS, Kimura S, Nalesnik MA, et al. The loss of renal dendritic cells and activation of host adaptive immunity are long-term effects of ischemia/reperfusion injury following syngeneic kidney transplantation[J]. Kidney Int, 2012, 81(10):1015-1025.
[40] Anzai A, Anzai T, Nagai S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling[J]. Circulation, 2012, 125(10):1234-1245.
[41] Santos-Zas I, Lemarié J, Tedgui A, et al. Adaptive immune responses contribute to post-ischemic cardiac remodeling[J]. Front Cardiovasc Med, 2018, 5:198.
[42] Rhee AJ, Lavine KJ. New approaches to target inflammation in heart failure: harnessing insights from studies of immune cell diversity[J]. Annu Rev Physiol, 2020, 82:1-20.
[43] Anzai A, Anzai T, Nagai S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling[J]. Circulation, 2012, 125(10):1234-1245.
[44] Nagai T, Honda S, Sugano Y, et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans[J]. J Am Heart Assoc, 2014, 3(3):e000839.
[45] Liu H, Gao W, Yuan J, et al. Exosomes derived from dendritic cells improve cardiac function via activation of CD4+T lymphocytes after myocardial infarction[J]. J Mol Cell Cardiol, 2016, 91:123-133.
[46] Lee JS, Jeong SJ, Kim S, et al. Conventional dendritic cells impair recovery after myocardial infarction[J]. J Immunol, 2018, 201(6):1784-1798.
[47] Van der Borght K, Scott CL, Nindl V, et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells[J]. Cell Rep, 2017, 18(12):3005-3017.
[48] Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+CD4+T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation[J]. Circ Res, 2014, 115(1):55-67.
[49] Saxena A, Dobaczewski M, Rai V, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function[J]. Am J Physiol Heart Circ Physiol, 2014, 307(8):H1233-H1242.
[50] Rymer JA, Newby LK. Failure to launch: targeting inflammation in acute coronary syndromes[J]. JACC Basic Transl Sci, 2017, 2(4):484-497.
[51] Adamo L, Staloch LJ, Rocha-Resende C, et al. Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury[J]. JCI Insight, 2018, 3(11):e120137.
[52] Choo EH, Lee JH, Park EH, et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization[J]. Circulation, 2017, 135(15):1444-1457.
[53] Hermansson A, Johansson DK, Ketelhuth DF, et al. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice[J]. Circulation, 2011, 123(10):1083-1091.
[54] 李大主,周游,吴伟,等. 负载热休克蛋白60的致耐受性树突状细胞疫苗对小鼠动脉粥样硬化斑块的影响[J]. 中国病理生理杂志, 2006, 22(6):1079-1082.
Li DZ, Zhou Y, Wu W, et al. Effects of rapamycin-treated HSP60-pulsed dendritic cells on the progression of the atherosclerotic plaque in mice[J]. Chin J Pathophysiol, 2006, 22(6):1079-1082.
[55] Frostegard J, Zhang Y, Sun J, et al. Oxidized low-density lipoprotein (OxLDL)-treated dendritic cells promote activation of T cells in human atherosclerotic plaque and blood, which is repressed by statins: microRNA let-7c is integral to the effect[J]. J Am Heart Assoc, 2016, 5(9):e003976.
[56] Zhou JJ, Wang YM, Lee VWS, et al. DEC205-DC targeted DNA vaccine against CX3CR1 protects against atherogenesis in mice[J]. PLoS One, 2018, 13(4):e0195657.
[57] Maekawa Y, Mizue N, Chan A, et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: a regulator of bone marrow-derived dendritic cells[J]. Circulation, 2009, 120(14): 1401-1414.
[58] Ma Y, Yuan J, Hu J, et al. ACE inhibitor suppresses cardiac remodeling after myocardial infarction by regulating dendritic cells and AT2 receptor-mediated mechanism in mice[J]. Biomed Pharmacother, 2019, 114:108660.
Role of dendritic cells in myocardial infarction and cardiac remodeling
ZHANG You-ming1, LIU hai-bo1,2△
(1,,,200120,;2,,,201700,)
Myocardial infarction (MI) is one of the leading causes of death worldwide. One of the primary reasons is that the rupture of atherosclerotic plaque leads to the formation of thrombosis, and then interrupts the coronary blood flow, thus finally causing the death of myocardial cells and cardiac dysfunction. A large number of researches have revealed that dendritic cells (DCs) play an essential role in immune inflammatory responses in the occurrence and development of atherosclerosis and MI. This article reviews the role of DCs in atherosclerosis, myocardial ischemia/reperfusion injury and cardiac remodeling after MI, and shows the potential values of DCs as an immunotherapeutic strategy for MI.
Dendritic cells; Myocardial infarction; Atherosclerosis; Ischemia/reperfusion injury; Myocardial remodeling
R542.2+1; R363.2
A
10.3969/j.issn.1000-4718.2020.11.025
1000-4718(2020)11-2093-06
2020-04-12
2020-05-21
国家自然科学基金资助项目(No.81770350)
Tel: 021-67009999; E-mail: haiboliu13@fudan.edu.cn
(责任编辑:宋延君,罗森)
猜你喜欢
今日农业(2022年14期)2022-09-15 01:43:28
摄影世界(2022年1期)2022-01-21 10:50:14
自我保健(2021年2期)2021-11-30 10:12:31
妇女之友(2021年9期)2021-09-26 14:29:36
昆明医科大学学报(2020年11期)2020-12-28 00:47:08
天津医科大学学报(2019年6期)2019-08-13 07:04:24
百姓生活(2019年2期)2019-03-20 06:06:16
知识经济·中国直销(2018年12期)2018-12-29 12:22:14
制造技术与机床(2017年6期)2018-01-19 02:41:10
商周刊(2017年6期)2017-08-22 03:42:36
