
Dysregulation of microRNA in cholangiocarcinoma identified through a meta-analysis of microRNA profiling
2020-11-30 06:53SomsakLikhitrattanapisalSupeechaKumkatePravechAjawatanawongKanokpanWongprasertRutaiwanTohtongTavanJanvilisri
World Journal of Gastroenterology 2020年29期
Somsak Likhitrattanapisal, Supeecha Kumkate, Pravech Ajawatanawong, Kanokpan Wongprasert, Rutaiwan Tohtong, Tavan Janvilisri
Abstract
Key words: Cholangiocarcinoma; Microarray; MicroRNA; Meta-analysis
INTRODUCTION
Cholangiocarcinoma (CCA), first described by Durand-Fardel in 1840, is a form of malignant tumor that originate from biliary epithelial cells in the liver and/or extrahepatic bile ducts[1,2]. CCA accounts for 10%-15% of hepato-biliary neoplasm. Thus, it is the second most common primary hepato-biliary malignancy after hepatocellular carcinoma (HCC)[3]. Moreover, the incidence and mortality rate of CCA have been reportedly increasing worldwide over the past three decades[2,4-6]. However, the prevalence of CCA vary greatly among different geographical regions of the world. Incidence of CCA in most Western countries ranges from 2 to 6 cases per 100000 people per year[7]. There is a higher prevalence of CCA in Asia and in people of Asian descent, which has been attributed to endemic chronic parasitic infestation[6,7]. Primary sclerosing cholangitis, inflammation that causes scars within the bile ducts, is the most common known predisposing factor for CCA[6]. In East and Southeast Asia, where the disease is common, CCA has been pathogenically associated with liver fluke infestation, particularly the endemicClonorchis sinensis[8]and Opisthorcis viverrini[9]. In addition, hepatitis C virus infection and liver cirrhosis have been suggested as potential risk factors for CCA[6].
MicroRNAs (miRNAs) are a family of endogenous, non-coding RNAs found in plants, animals, and some viruses[10-13]. miRNA genes are highly-conserved and may be located either within the introns or exons of protein-coding genes (70%) or in intergenic areas (30%)[10,14]. A miRNA in its single-stranded functional form is usually 21-22 nucleotides long (though it can vary from 19-25 nucleotides)[10,13]. In present, the latest release of miRBase (version 22) (http://www.mirbase.org/) contains 38589 hairpin precursors and 48860 mature miRNA from 271 organisms[15]. Several hundreds of miRNA genes in the human genome have been discovered in the last decade[10,13,16]. However, it is estimated that the human genome may encode over 1000 miRNAs in total[17,18]. The primary function of miRNAs is to control gene expression at posttranscriptional level. miRNAs suppress the target mRNA expression, mostly through interaction with the 3’ untranslated region[13,19], resulting in inhibition of target mRNA translation activity and, to a lesser extent, targeting mRNA cleavage[11,16,20]. Each miRNA may be responsible for regulation of the expression of hundreds of gene targets[19].
Although the functions of dysregulated miRNAs in human cancers remain largely a mystery, multiple miRNAs and their corresponding target genes have been reported to be associated with tumor initiation and progression[18,21,22]. Many transcriptional profiling data demonstrated that miRNA expression profiling efficiently classified different tumor types more reliably than did mRNA profiling[23,24]. Systematic expression analyses using miRNA microarray technology have been performed in several types of cancer; for example, hepatic[25], colorectal[26], lung[27], and breast cancers[28].
However, due to insufficient control of false positives and the small sample sizes relative to the large sets of microarray probes, individual microarray-based studies are often deficient in terms of statistical robustness[29,30]. Meta-analysis is the use of statistical techniques to combine results from independent but related studies, hence it is one of the most preferable ways to increase the statistical power of the readily available microarray data. Moreover, it is relatively inexpensive in terms of financial and time investments[31].
Our meta-analysis of miRNA microarray datasets was aimed to identify the differentially expressed (DE) miRNA in various CCA samples compared to the noncancerous counterparts. The results from the robust statistical tests would provide insights to understanding the regulatory potential of miRNAs in tumorigenesis pathways of CCA.
MATERIALS AND METHODS
miRNA microarray data collection
The miRNA microarray datasets were retrieved from the public repository database Gene Expression Omnibus (http: //ncbi.nlm.nih.gov/geo)viathe computerized search using combinations of relevant keywords, including (miRNA OR microRNA) AND (cholangiocarcinoma OR CCA OR CCC). All dataset search hits were initially checked whether raw data were provided. Datasets without raw data were promptly omitted. A matrix series tables and raw data package(s) of each available dataset were downloaded and extracted. Eight miRNA microarray datasets from independent research studies were employed in our meta-analysis. Six out of these 8 datasets (GSE32958, GSE47764, GSE50894, GSE51429, GSE53870, GSE53992) were conducted using biopsied tissue samples. One study (GSE59856) used serum miRNA and another (GSE47396) used miRNA samples from cell lines.
Data processing
After extraction and background-correction, the GPR or TXT raw data files of each dataset were converted to comma-separated values file format and then imported as a data frame into R software version 3.1.2[32]. Before pooling samples of all datasets, the identification name of every miRNA probe was checked against identification entries registered in miRBase database (www.mirbase.org/)[15]and was subsequently renamed in accordance with miRBase identification, in order to avoid confusion from naming system of different microarray platforms. Log2transformation was applied to all intensity values. The transformed data were then normalized following 2 steps,i.e., within-array normalization using a median centering method, followed by betweenarray normalization using a quantile normalization method. Hence normalized data would exhibit normal distribution with the same standard deviation across datasets which is an essential assumption of parametric statistical tests.
Statistical analysis
In order to identify DE miRNA in CCA, the pooled samples were grouped into 2 groups: “CCA” and “Normal”. The normalized intensity values of each miRNA were compared between these two groups usingt-tests conducted in MultiViewer Experiment version 4.6 in TM4 software suite[33].Pvalue threshold was set at below 0.001 (P< 0.001). As clinical validation in our study is restricted,k-fold cross validation was applied to verify the integrity of significant DE miRNAs fromt-test analysis. The 10-fold cross validation was performed by randomly assigning samples into 10 different sets and repeatingt-test statistical analysis with the same parameter (P< 0.001) for 10 rounds. In each round, one set was chosen as the validation set whereas the rest were test sets. DE miRNA which showed statistical significance among test sets and validation sets from all 10 rounds were collected as validated DE miRNAs for further analysis.
Bioinformatics analysis and visualization of miRNA-target interactions
In order to assess the biological functions of the gene targets of DE miRNAs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were employed using DIANA miRPath version 3.0[34]. The lists of up- and down-regulated miRNAs were categorized and separately uploaded as inputs to DIANA miRPath. The human KEGG and GO analyses were selected with P-value threshold at 0.01. The KEGG pathways, which were the most enriched by target genes of DE miRNAs, were selected to analyze the miRNA-target interactions. Unique targets of each DE miRNA in the selected pathways were filtered into the list, which was then used in the network visualizer software. Visualized miRNA-target interaction networks were constructed using Cytoscape version 3.3.0[35]with CyTargetLinker plugin version 2.1[36].
RESULTS
Differentially-expressed miRNA
A framework of our meta-analysis approach is depicted in Figure 1. In total, there were eligible 246 CCA and 197 normal samples from 8 independent miRNA microarray datasets, which included tissue samples, sera and cell lines as detailed in Table 1. Initialt-tests results identified 224 DE miRNAs consisting 114 up-regulated and 110 down-regulated miRNAs. Following statistical validation of these results usingk-fold cross validation, the numbers of up-regulated and down-regulated miRNAs were confined to 70 (Table 2) and 48 (Table 3), respectively. The overall expression profiling of DE miRNAs is presented in Figure 2.
Enrichment analyses
To explore the biological functions of DE miRNAs found in the meta-analysis results, GO and KEGG pathway enrichment analyses were performed using DIANA miRPath v3.0 withPvalue threshold at 0.01 and MicroT score threshold at 0.8. The miRPath v.3.0 results indicated that there were 4407 and 7236 predicted target genes of upregulated and down-regulated miRNAs, respectively, involved in GO biological processes. GO enrichment analysis showed 72 biological processes associated with upregulated miRNAs (Supplementary Table 1), whereas 95 biological processes were identified to be associated with down-regulated miRNAs (Supplementary Table 2).
KEGG pathway enrichment analysis showed that gene targets of up-regulated miRNAs were significantly involved (P< 0.01) in 48 molecular biological processes (Supplementary Table 3) while gene targets of down-regulated miRNAs were significantly involved (P< 0.01) in 32 processes (Supplementary Table 4). Figure 3 represents the most prominent dysregulated pathways of up-regulated miRNAs including phosphatidylinositol-3 kinases/Akt (PI3K/Akt) signaling pathway (215 gene targets of 57 miRNAs,P= 0.00424), mitogen-activated protein kinase (MAPK) signaling pathway (169 gene targets of 55 miRNAs, P = 0.00034), and Ras signaling pathway (159 gene targets of 58 miRNAs,P= 2.34E-06). These three pathways were also the most prominent dysregulated pathways of down-regulated miRNAs, which were PI3K/Akt signaling pathway (209 gene targets of 42 miRNAs,P= 1.91E-05), MAPK signaling pathway (147 gene targets of 39 miRNAs,P= 0.0079), and Ras signaling pathway (147 gene targets of 39 miRNAs,P= 2.03E-06).
Visualization of miRNA-target interaction networks
According to the results of KEGG pathway analysis, PI3K/Akt, MAPK and Ras signaling pathways were the most enriched biological processes with which target genes of DE miRNAs were associated. For up-regulated miRNAs associated with these three pathways, 7 miRNAs including miR-330-5p, miR-519d-3p, miR-548a-5p, miR-548d-3p, miR-1207-3p, miR-1304-5p, and miR-2113 were chosen as representative miRNAs of this group. For down-regulated miRNAs, 9 miRNAs including let-7b-5p, let-7c-3p, let-7f-5p, miR-195-5p, miR-20a-5p, miR-26b-5p, miR-27b-3p, miR-29b-3p, andmiR-330-3p were chosen as representatives. Using regulatory interaction networks data from TargetScan and miRTarBase, 35 and 46 unique target genes of up- and down-regulated miRNAs, respectively, were identified to be involved in these three pathways (Figure 4).

Table 1 Summary of microRNA microarray datasets used in this study
DISCUSSION
At present, a global view of miRNA roles in the development of cancers remains incomplete. Due to widespread usage of microarray technology, there has been an enormous expansion of publicly available datasets[37], which could be integrated and analyzed with a statistically robust meta-analysis approach. In our meta-analysis, miRNA microarray datasets from multiple independent studies were analyzed with highly stringent statistics and cross validation, leading to identification of DE miRNAs in CCA compared to non-cancerous cells across the pooled samples. The lack information on whether the samples were from primary or metastatic sites would pose as one of the limitations of this study.
Many DE miRNAs observed in our study are significantly related to 3 major cancer signaling pathways, namely the MAPK signaling pathway, PI3K/Akt signaling pathway, and Ras signaling pathway. Among the up-regulated miRNAs found in our meta-analysis, miR-519d and miR-330 are of particular interest as dysregulated expression of these miRNAs have been reported in several types of cancer. miR-519d belongs to the chromosome 19 miRNA cluster, which is the largest human miRNA cluster described so far[17]. Although there is no direct evidence on the relationship of miR-519d and CCA development, miR-519d has been shown to be up-regulated in HCC patient’s tissues, exerting oncogenic activity by inhibiting the tumor suppressor proteins such as CDKN1A/p21, PTEN, AKT3 and TIMP2[38]. In contrast, overexpression of miR-519d in a human HCC cell line QGY-7703 has been shown to block cell proliferation. On the contrary, down-regulation of miR-519d has been reported to promote cell proliferation in many cancers, including cervical cancer, breast cancer, and ovarian cancer[39-42].
Another up-regulated miRNA identified in our study includes miR-330. This miRNA has been previously reported to be up-regulated in glioblastoma[43], colorectal cancer[44], non-small cell lung cancer[45], and esophageal cancer[46]whereas its downregulated expression has been demonstrated in prostate cancer[47-49]. miR-330 has been shown to promote cancer cell proliferationviasuppression ofCDC42, a Rho GTPaseassociated with MAPK signaling pathway[44]. Besides, hypoxia-induced upregulation of integrin-alpha 5, a predicted target of miR-330 and a critical receptor in PI3K/Akt signaling pathway, has been shown to enhance cell proliferation, metastasis and apoptosis resistance of CCA cell lines[50-52]. Paradoxically, miR-330 has been reported to induce apoptosis in prostate cancer cells through E2F1-mediated suppression of Akt phosphorylation in prostate cancer cells[47].
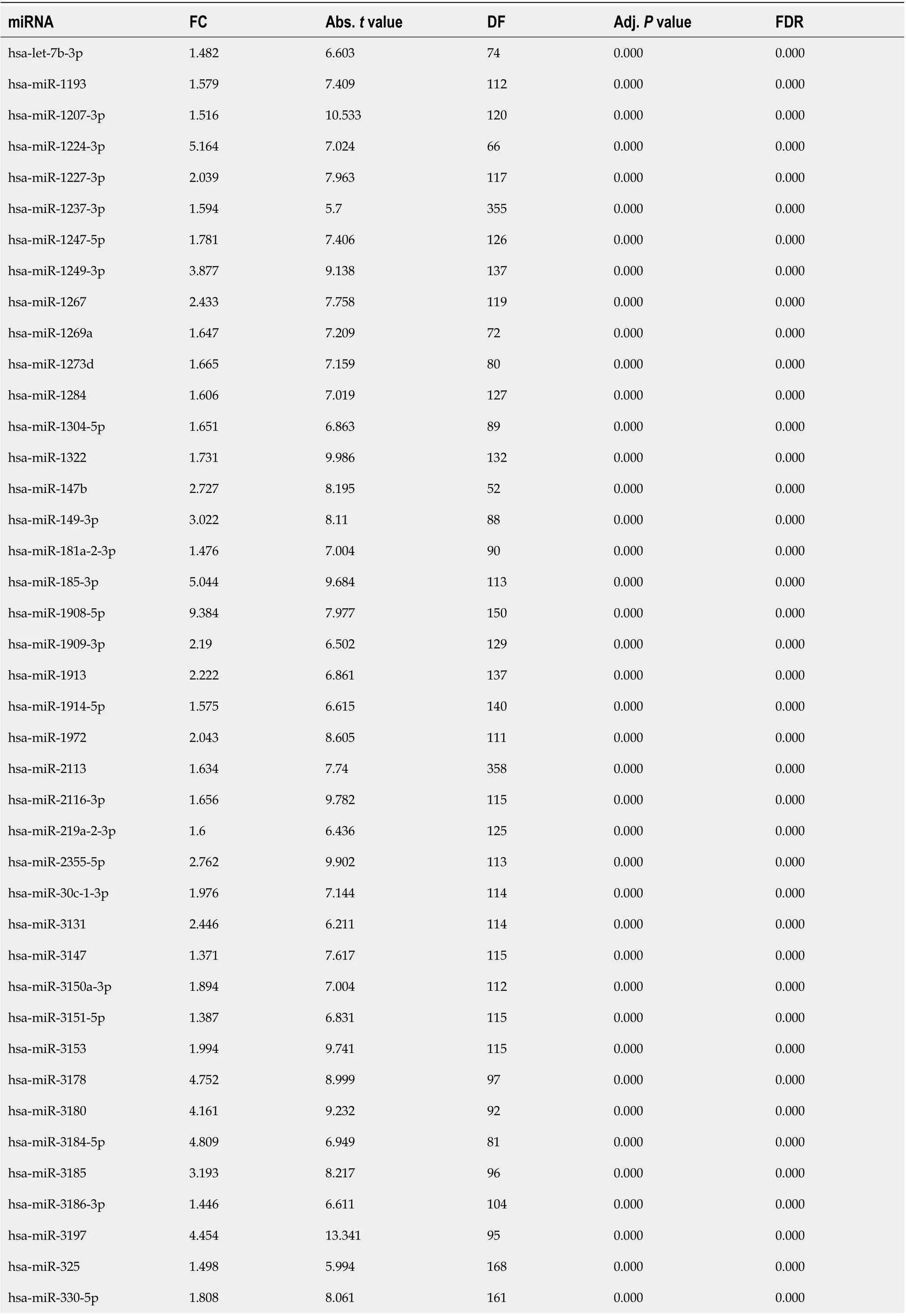
Table 2 List of 10-fold-cross-validated differentially-expressed microRNAs which were up-regulated in cholangiocarcinoma

The P value was set to lower than 0.001 (P < 0.001). miRNA: MicroRNA; FC: Fold change (ratio of mean signal intensities of cholangiocarcinoma to those of normal samples); DF: Degree of freedom; Abs. t value: Absolute t value; Adj. P value: Adjusted P value; FDR: False discovery rate.
One of the down-regulated miRNAs identified in our study was miR-20a, one of the mature miRNA products of the miR-17-92 cluster pri-miRNA. Down-regulation of miR-20a has been shown to mediate cellular differentiation and growth arrest induced by HIF-1 in acute myeloid leukemia cells by targeting p21 and STAT3[52], demonstrating an oncogenic role of miR-20a. In addition, the miR-17-92 cluster miRNAs have been shown to be up-regulated and play oncogenic roles in many types of cancer including CCA[53]. In contrast, down-regulation of miR-20a has been shown to promote HCC cell proliferationviaupregulation of Mcl-1, an antiapoptotic member of the Bcl-2 family, suggesting a tumor-suppressor role[54].
Besides, our meta-analysis of CCA miRNA microarrays has revealed downregulation of several members of the let-7 miRNA family (let-7b, -7c, -7f), whose members are estimated to comprise 1%-5% of the mammalian genome[10,17]. This miRNA family has been shown to generally play a tumor suppressor role[55], where ectopic expression of the let-7 family inhibits cell proliferation through downregulation of c-Myc in nasopharyngeal carcinoma cells, whereas down-regulation of let-7 promotes cancer cell growth by increasing the activity of Ras protein, in lung cancer[56]and liver cancer[57]cells.

Table 3 List of 10-fold-cross-validated differentially-expressed microRNAs which were down-regulated in cholangiocarcinoma
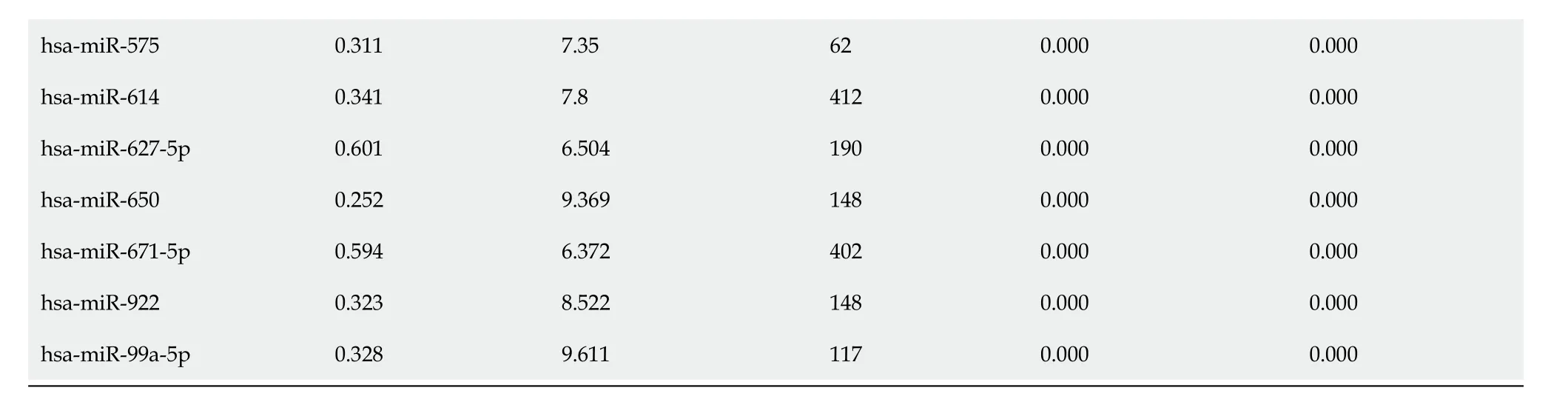
The P value was set to lower than 0.001 (P < 0.001). miRNA: MicroRNA; FC: Fold change (ratio of mean signal intensities of cholangiocarcinoma to those of normal samples); DF: Degree of freedom; Abs. t value: Absolute t value; Adj. P value: Adjusted P value; FDR: False discovery rate.
Altogether, the meta-analysis of miRNA microarray datasets with highly stringent statistical methodology provides new insights into the role of miRNA and its dysregulations in CCA. Our findings of miRNA dysregulations in the cancer signaling pathways including PI3K/Akt pathway, MAPK pathway, and Ras pathway give clues into underlying miRNA-mRNA interplays of CCA. However, the analyses reported herein were based on the different origins of miRNAs, validations of such findings are warranted. Of clinical relevance, since many miRNAs have been reported as highly specific biomarkers for several types of cancer[58-60], the identified miRNA in this study may have predictive values for CCA cases. Also, there are conflicting results in adjuvant settings for CCA[61], the detection of a specific miRNA may be associated with an increased risk of recurrence after surgery. Further investigation of the miRNAs reported herein will bring about the novel knowledge of the dysregulated processes in CCA development at post-transcriptional level which could offer novel diagnostic and therapeutic approaches in the future.
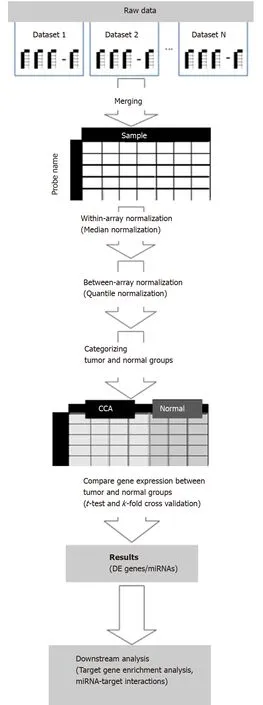
Figure 1 Overview of a meta-analysis approach in this study. CCA: Cholangiocarcinoma; DE: Differentially expressed; miRNA: MicroRNA.

Figure 2 Heatmap of microRNA expression in cholangiocarcinoma and normal samples. The color gradient of each cell represents the log2 of normalized intensity value of microRNA microarray spot. CCA: Cholangiocarcinoma; miRNA: MicroRNA.

Figure 3 Pathway enrichment analyses of predicted target genes of differentially expressed microRNAs. The differentially expressed microRNAs obtained from meta-analysis were input to DIANA miRPath version 3.0. P value thresholds were set at 0.01. PI3K: Phosphatidylinositol-3 kinases; MAPK: Mitogen-activated protein kinase.

Figure 4 MicroRNA-target interaction networks. A: Up-regulated; B: Down-regulated. MicroRNA (miRNA)-target interaction networks of 7 up-regulated and 9 down-regulated miRNAs associating in phosphatidylinositol-3 kinases/Akt, mitogen-activated protein kinase, and Ras signaling pathways based on pathway enrichment analysis via DIANA miRPath version 3.0. Blue and red lines indicate the miRNA-target prediction or information based on TargetScan and miRTarBase databases, respectively. PI3K: Phosphatidylinositol-3 kinases; MAPK: Mitogen-activated protein kinase.
ARTICLE HIGHLIGHTS

Research objectives
This work integrates and intervalidates the CCA miRNA expression profiles from multiple independent datasets to identify the differential dysregulation of miRNA and their corresponding downstream pathways underlying mechanism of pathogenesis.
Research methods
Eight independent CCA miRNA profiling microarray datasets, including 246 CCA and 197 normal samples were assimilated into a meta-analysis and cross-validation to identify a cohort of miRNA that were significantly dysregulated in CCA.
Research results
Of 118 dysregulated miRNA identified in our study, 70 were up-regulated and 48 were down-regulated miRNAs in CCA. Bioinformatic analyses revealed that mRNA targets of differentially expressed miRNAs were significantly distributed across various biological processes. The most prominent dysregulated pathways included phosphatidylinositol-3 kinases/Akt, mitogen-activated protein kinase and Ras signaling pathways.
Research conclusions
This current study represents the meta-analysis of miRNA microarray datasets with highly stringent statistical methodology and provides new insights into the role of miRNA and its dysregulations in CCA.
Research perspectives
The merit of our findings offers a valuable reference for future studies and further investigation of these miRNA/genes and their interactions will eventually lead to the identification of genes and pathways important to the overall mechanism of the dysregulated processes in CCA development.
ACKNOWLEDGEMENTS
We appreciate a scholarship from the Development and Promotion of Science and Technology Talented Project awarded to Likhitrattanapisal S.
 World Journal of Gastroenterology2020年29期
World Journal of Gastroenterology2020年29期
- World Journal of Gastroenterology的其它文章
- Endoscopic management of gastrointestinal leaks and fistulae: What option do we have?
- Watch and wait approach in rectal cancer: Current controversies and future directions
- Evaluation of intrahepatic manifestation and distant extrahepatic disease in alveolar echinococcosis
- Multivariate predictive model for asymptomatic spontaneous bacterial peritonitis in patients with liver cirrhosis
- Clinicopathological characteristics and surgical outcomes of sarcomatoid hepatocellular carcinoma
- Patients' perspectives on smoking and inflammatory bowel disease: An online survey in collaboration with European Federation of Crohn's and Ulcerative Colitis Associations