
Profi le and development of microsatellite primers for Acanthogobius ommaturus based on high-throughput sequencing technology*
2020-11-26 08:41:06SONGChenyuFENGZiyiLIChunhouSUNZhichengGAOTianxiangSONGNaLIULu
SONG Chenyu , FENG Ziyi , LI Chunhou , SUN Zhicheng , GAO Tianxiang , , SONG Na , , LIU Lu
1 Fisheries College, Ocean University of China, Qingdao 266003, China
2 South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou 510300, China 3 National Engineering Research Center for Marine Aquaculture, Zhejiang Ocean University, Zhoushan 316022, China
Abstract Acanthogobius ommaturus, a fi sh species of the Family Gobiidae, is a marine commercial fi sh perched on the bottom of seawater. In this study, Illumina high-throughput sequencing technology was applied to obtain the candidate microsatellite markers of A. ommaturus. A total of 4 746 microsatellite-rich fragments were found, of which 4 542 microsatellites are with primer fragments, containing 971 dinucleotide sequences, 2 643 trinucleotide sequences, 569 tetranucleotide sequences, 406 pentanucleotide sequences, and 212 hexanucleotide sequences. Based on the results of high-throughput sequencing, a total of 141 pairs of the microsatellite primers were designed and screened. And then 24 polymorphic primers were fi nally obtained by polyacrylamide gel electrophoresis. In total, 271 alleles were detected in the 24 pairs of primers. The number of alleles for diff erent primers ranged from 5 to 19. The average number of eff ective alleles ( N a) was 11.292; the average observed heterozygosity ( H o) of the 24 pairs of primers was 0.665, the average expected heterozygosity ( H e) was 0.880, and the average polymorphic information content was 0.846. All sites were highly polymorphic (PIC>0.50).
Keyword: microsatellite; Acanthogobius ommaturus; high-throughput sequencing; polymorphic sites
1 INTRODUCTION
Microsatellite or simple sequence repeats (SSR) is a simple tandem repeat consisting of a few nucleotides (generally 1–6 repeat units) (Zane et al., 2002). The length polymorphism of these sequences is due to diff erent repetition times and inconsistent repeat units. Microsatellites are widely distributed in the genomes of eukaryotes and prokaryotes. Microsatellite molecular marker technology is characterized by high abundance and reproducibility, codominant markers, and neutral selection (Zane et al., 2002; Zhang and Hewitt, 2003; Li, 2004). As a co-dominant molecular marker, microsatellite marker has the advantages of rich polymorphism, wide distribution range, high stability and easy detection. It has been widely used in various fi elds of conservation genetics, including population genetic diversity assessment, population genetic structure analysis, paternity testing etc. (Arens et al., 2006; Uller et al., 2006; Barbará et al., 2007; Castoe et al., 2010).
Acanthogobiusommaturus, belonging to Osteichthyes, Perciformes, Gobioidei, Gobiidae, is a demersal fi sh that is widely distributed in coastal areas and estuaries of China, North Korea, Japan, and Indonesia. There are several formerly used names:Synechogobiushasta,AcanthogobiushastaandSynechogobiusommaturus(Zhao and Wu, 2008). Although microsatellite marker has been widely used in the studies on population genetics of marine organisms (Winkelmann et al., 2013), no population genetic research has been conducted on theA.ommaturusuntil now. Studies onA.ommaturusmainly focused on the comparative analysis of individual fecundity and morphology (Feng et al., 2004), fi shery biology (Fan et al., 2005), name of species (Song et al., 2010), DNA barcode (Gu et al., 2013), and genetic diversity and structure by other molecular markers (Song et al., 2010a, b, 2011). Bai et al. (2012) used magnetic bead enrichment method to develop 17 pairs of microsatellite primers. The number of microsatellite markers forA.ommaturusis far from enough for a more comprehensive population genetic analysis. High-throughput sequencing technology allows large-scale, accurate, inexpensive, and rapid sequencing of species (Holt and Jones, 2008). By analyzing the characteristics of the sequencing results in detail, we can provide a stronger basis for the following development ofA.ommaturusprimers to analyze the characteristics of microsatellite markers, screen more microsatellite primers, and lay foundation for further studies on the genetics and conservation of this species.
2 MATERIAL AND METHOD
2.1 Sampling and DNA extraction
OneA.ommaturussample was collected from Laizhou Bay (Fig.1a), China, and sent for highthroughput sequencing. Twenty-four individuals ofA.ommaturuswere collected from the southern coastal waters near Xiamen (Fig.1b), China, and were used for polymorphism detection and genetic diversity analysis. These samples were preserved in 95% alcohol at the -20°C for DNA extraction. Genomic DNA was extracted from muscle tissue with the standard phenol-chloroform method described by Sambrook and Russell (2001).
2.2 High-throughput sequencing
DNA samples were detected by agarose gel electrophoresis, and the purity and integrity of DNA were analyzed. DNA samples with clear bands and no tail conform to the requirements of database construction. A microsatellite-enriched genomic library was constructed following the enrichment protocols of Ma and Chen (2009). Qualifi ed DNA samples were digested and randomly interrupted. The preparation of the whole library needs terminal repair, A tail addition, sequencing connector addition (P5: 5′- AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT-3′; P7: 5′-TCGGAAGAGCACACGTCTGAACTCCAGTCACGCAGGAATCTCGTATGCCGTCTTCTGCTTG- 3′), purifi cation, PCR amplifi cation, recovery of 300–700 bp sequence and other steps to complete. In the end, the constructed genomic library were sequenced by Illumina HiSeqTM2000 (Illumina, USA) in the biological company (Personal Biotechnology Co., Ltd., Shanghai).
2.3 Acquisition of SSR sequence
The original sequences were clustered and assembled, and then the microsatellite sequences were detected by SSR search software. The standard of isolating SSR are as follows. The minimum length of repeating motif is 2 bp and the maximum length is 6 bp; the minimum length of the SSR sequence is 12 bp; The SSR upstream and downstream sequence length is 100 bp; the minimum distance between two microsatellite DNAs is 12 bp.
2.4 The detection of primer polymorphism
One hundred and forty-one PCR primer pairs were preferred to be designed using the PRIMER PREMIER 5 software (Premier Biosoft International, USA). The conditions are: repetition times of SSR units are more than 6; length of SSR units is 3–10 bp; expected length of PCR products is 130–300 bp; primers that repeat the same four bases continuously are excluded.
The polymorphism of these primer pairs were tested using 24A.ommaturussamples. The PCR were conducted in volumes of 25 μL reaction mixtures containing 17.25-μL ultrapure water, 2.5-μL 10× PCR buff er, 2-μL dNTPs, 1-μL each primer (5 μmol/L), 0.25-μL Taq polymerase, and 1-μL DNA template. The PCR program was as follows: 5 min for denaturation at 95°C, followed by 35 cycles of 45 s at 94°C, 1 min at the annealing temperature (Table 1), 45 s at 72°C, and a fi nal step for 10 min at 72°C. Then the PCR products were held at 4°C for insulation. After the amplifi cation, the PCR products were electrophoresed on 8% non-denaturing polyacrylamide gel at 14 W for 3–4 h and visualized by silver staining (Lin et al., 2011). Allele size was identifi ed according to the 20-bp DNA ladder (TaKaRa, China).
2.5 Data analysis
After statistical analysis of electrophoresis results, the results were input into the population genetic analysis software Genepop 4.0 (Rousset, 2008). The related parameters of SSR primers were calculated, including the average number of eff ective alleles (Na), polymorphic information content (PIC), apparent heterozygosity (Ho) and expected heterozygosity (He), and Hardy-Weinberg equilibrium test and linkage disequilibrium test were carried out.
3 RESULT
3.1 Raw data quality
Raw reads 6.606 G was born and clean reads 6.511 G was fi ltered. The raw reads of each sequence were 6 606.008 M, with high quality (Q20 ≥ 95.6%, Q30 ≥ 93.92%). The GC content was 39.39%, and the RAD-Tag capture rate was 97.33%. Sequences were then spliced and assembled. The total contig base was 96 594 144 bp, the total contig number of the sequences was 320 503, the average contig length of the assembly sequence was 301 bp, and the GC content of assembly result was 39.01%. Reads after de-duplication are compared to the assembly results. Variation detection showed that most of the heterozygous SNPs (Het rate (%): 83.73) and a few homozygous SNPs appeared in the test results, indicating that the assembly results were reliable.
3.2 Characterization of microsatellite markers
There are 4 756 microsatellite-rich fragments were examined to reduce redundancy and the number of microsatellites with primer fragments is 4 542. A total of 971 reads contained dinucleotide repeats, 2 643 contained trinucleotide repeats, 569 contained tetranucleotide repeats, 146 contained pentanucleotide repeats and 212 contained hexanucleotide repeats (Fig.2). The number of trinucleotide repeats was the highest. The number of eleven nucleotides was the lowest. Ttrinucleotide repeats had the greatest percentage of SSR mutation type distribution (Fig.2).
The AT repeat motifs was the most frequent among all ten types of dinucleotide repeats, whereas CT was the least frequent (Fig.3a). The ATT repeat motifs were the most frequent among all fi fty-six types of trinucleotide repeats (Fig.3b). The CATG, AAAT, AATG repeat motifs were the three most frequent tetranucleotide motifs (Fig.3c), and 96% of all tetranucleotide motifs were less than 30 repeats. TheAATTG repeat motifswere the most frequent pentanucleotide (Fig.3d) and TTCTGA was the most frequent hexanucleotide (Fig.3e).
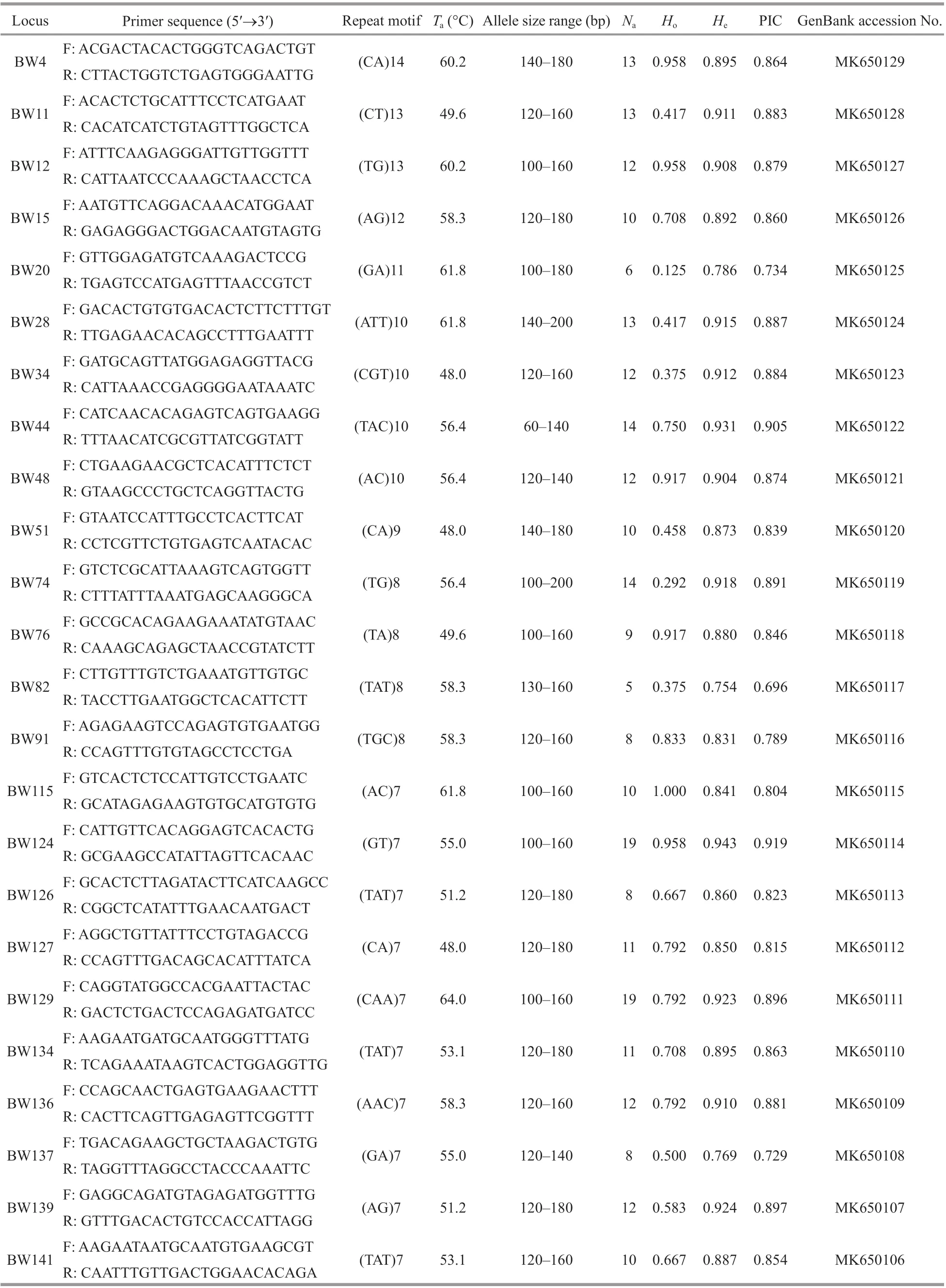
Table 1 Characteristics of microsatellite loci in A. ommaturus
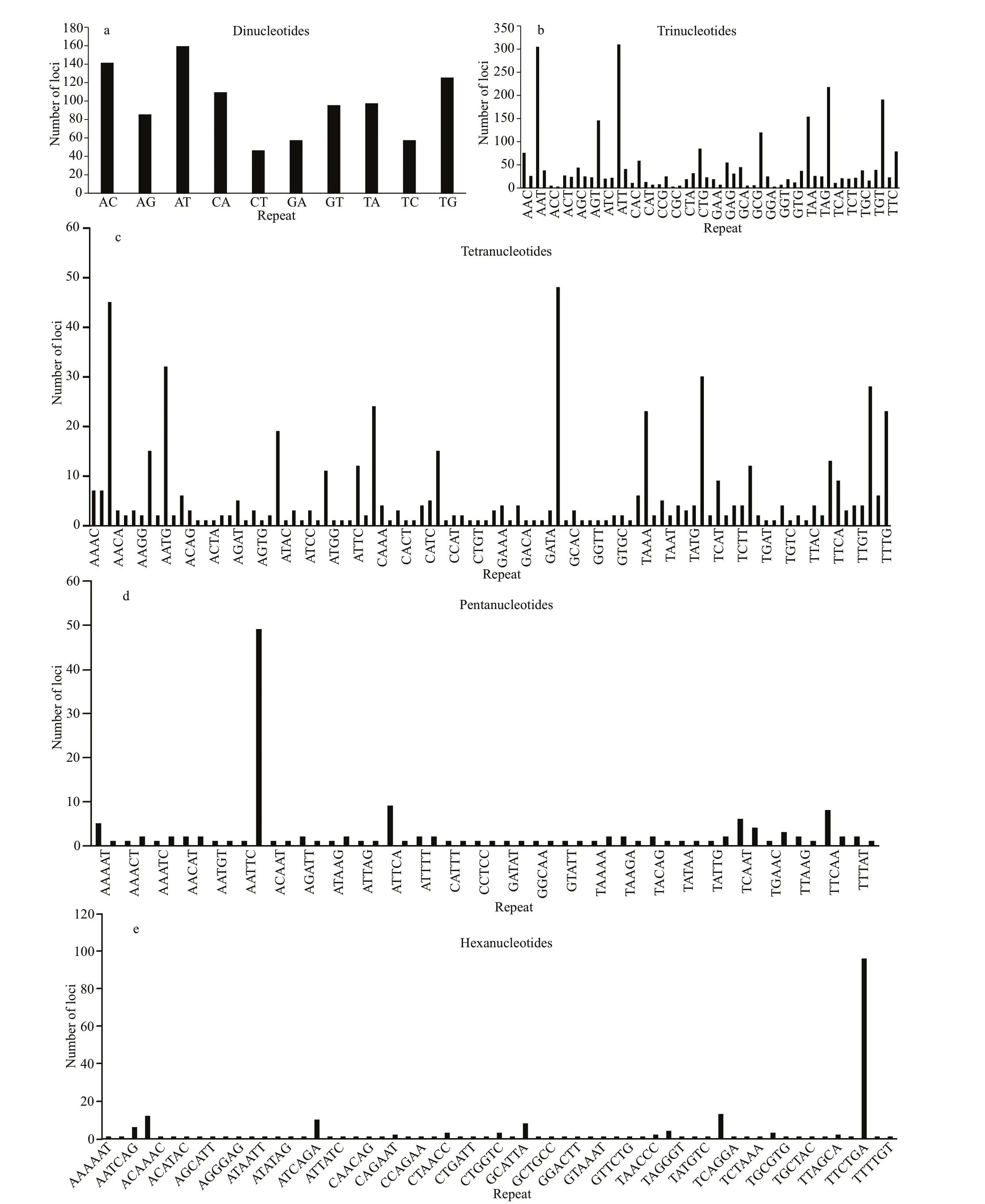
Fig.3 Type and number of repeat motif for dinucleotide (a), trinucleotide (b), tetranucleotide (c), pentanucleotide (d), and hexanucleotide (e)
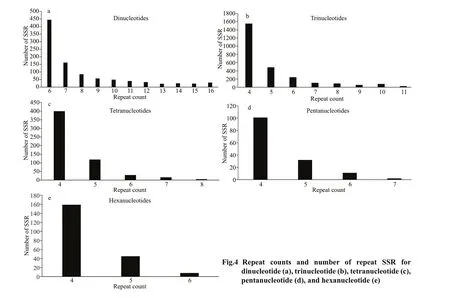
The number of SSRs in the dinucleotide type with 6 repeat units corresponds to an absolute dominant position, which is 45.83% of the total SSR (Fig.4a). Trinucleotide-type SSRs have the highest number of repeats 4 with repeat units, accounting for 58.76% (Fig.4b). Tetranucleotide-type SSRs have the highest number of 4repeats units, accounting for 70.30% (Fig.4c). The number of SSRs with 4 pentanucleotide type repeats was the highest, accounting for 69.18% (Fig.4d). The number of SSRs with the hexanucleotide type repeating sequence repetition number 4 is the highest, accounting for 75.00% (Fig.4e).
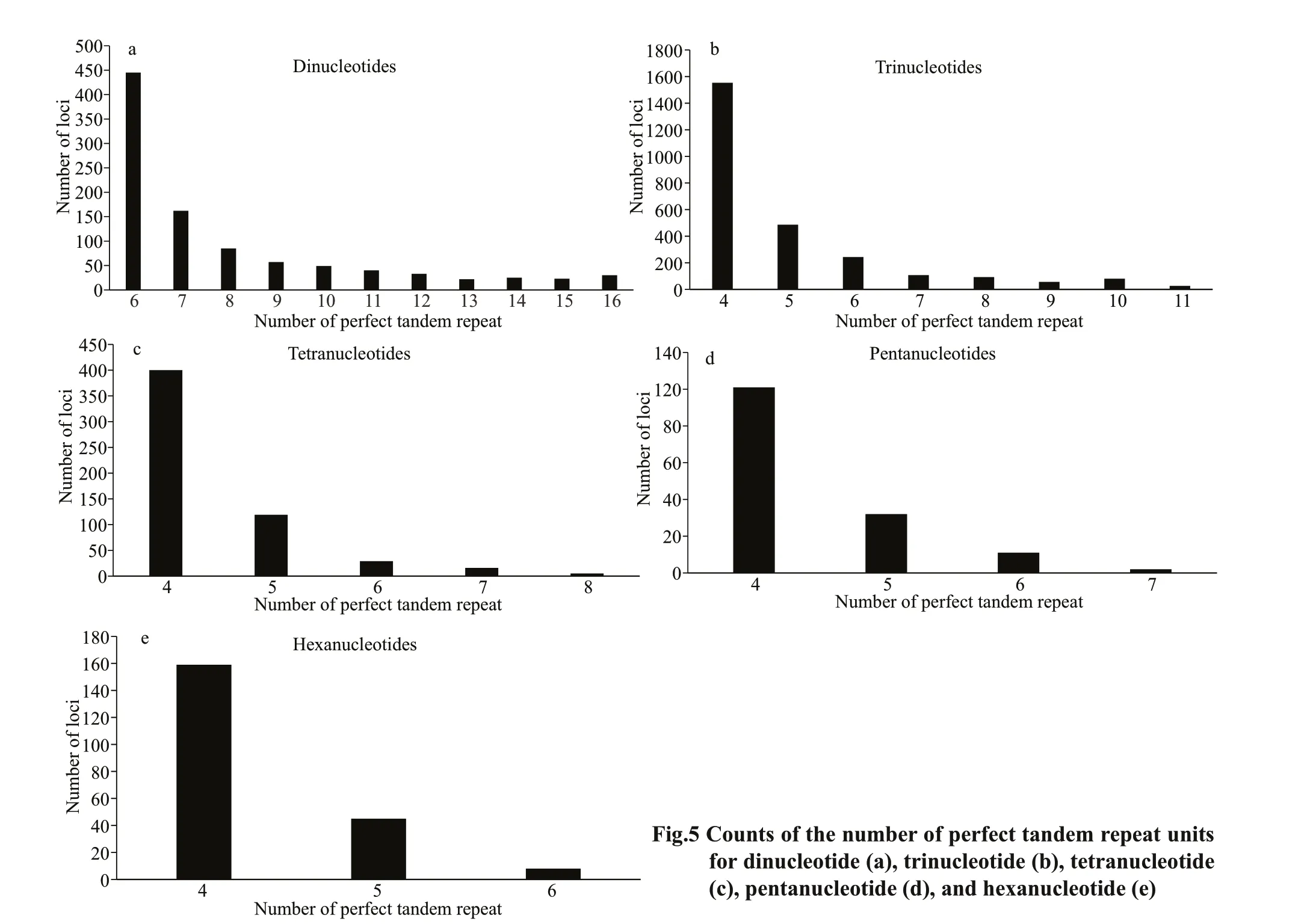
The number of dinucleotide repeats is 6–16, the repeat number of trinucleotides is 4–11, the repeat number of tetranucleotides is 4–8, the repeat number of pentanucleotides is 5–7, and the number of hexanucleotide repeats is 4–8, with a signifi cant downward trend. As the number of perfect tandem repeat increased, the number of loci decreased (Fig.5). The perfect trinucleotide tandem repeat had the greatest numbers of loci (Fig.5b).
3.3 Detection of primer polymorphism
One hundred and forty-one primer pairs were tested by agarose gel electrophoresis and 24 primers showed polymorphic. The number of alleles per locus ranged from 5 to 19, with an eff ective average of 11.292. The observed (Ho) and expected (He) heterozygosities varied from 0.125 to 1.000 and from 0.754 to 0.943, respectively (Table 1). Eleven out of 24 polymorphic loci were signifi cantly deviated from Hardy-Weinberg equilibrium (P<0.05). The polymorphism information content ranged from 0.696 to 0.919, all loci were showed highly polymorphic. Five loci (BW20, BW34, BW44, BW74, and BW129) were shown evidence of null alleles (null alleles frequency>5%). No genetic disequilibrium was detected in each locus.
4 DISCUSSION
4.1 Raw data quality
According to the sequencing results, the data quantity is good, the quality of sequencing is qualifi ed, the GC distribution is normal, and the database is successfully constructed and sequenced (Zerbino and Birney, 2008). Yang et al. (2015) ever sequenced the genome ofParargyropseditaby random highthroughput sequencing, and the results show that the original sequence data were 2.80 Gbp. The diff erence of results may be caused by the diff erent parameters of search software and software settings or the diff erent genome size. The accuracy of assembly results can be refl ected by the consistency between the GC content of assembly results and that of reads. The GC content of the assembly results is consistent with that of the sequencing data, which can refl ect that the assembly results can represent part of the genome (Wang et al., 2017; Gao et al., 2018). The rate of designing fragment primer is 95.5%. To sum up, compared with traditional methods, high throughput sequencing technology can capture more microsatellite fragments more accurately in a shorter time.
4.2 Characterization of microsatellite markers
The number of repeated units tandemly in microsatellites is diff erent from other species, which may be the result of natural selection and gene mutation (Katti et al., 2001). The results of the highest number of trinucleotide repeat units is consistent with other previous studies, such as inCrassostreagigas(Zhang et al., 2012), corals (Wang et al., 2009). The number of repeats decreases exponentially with repeat unit length in most eukaryotes because of higher gene mutation in longer repeats (Katti et al., 2001). It is reported that the number of repeats is negatively correlated with repeat length (Chen et al., 2010). The results of this study follow thispattern.
Some studies believe that the number of dinucleotide repeat unit of vertebrates is the most abundant (Wang et al., 2012). Therefore, this type of microsatellite loci has been widely used in molecular ecology, and AC repeat units are more commonly found (Tόth et al., 2000; Chistiakov et al., 2006). For example, the largest number of AC repeat units was found in the genome ofPatinopectenyessoensis(Guo, 2014). The AG is the most common dinucleotide repeat unit in theExopalaemoncarinicaudamicrosatellite core sequence (Duan et al., 2016). The largest number of repeat units was AT in this study, which was diff erent from common distributions. Furthermore, the ATG (Guo, 2014) and the AAT (Duan et al., 2016) is the most common trinucleotide repeat unit in the genome ofExopalaemoncarinicauda, while the types of trinucleotide repeats are diff erent in the present study. Most of this result depends on the diff erences in species genomes, but it may also be related to the construction of cloned libraries and the length of sequencing fragments (Guo, 2014).
We found that the number of SSRs with a core sequence repeat number of four accounts for an absolute dominant position, followed by 6. However, the number of these two SSRs types is quite diff erent. The number of SSR decreased with the increase of repetition times. The number of copies of the repeat unit and the number of microsatellite sequences are negatively correlated. The concentrated region of SSRs in genome may be the region where organisms respond rapidly under diff erent environmental pressures (Li et al., 2004; Kashi and King, 2006). The organism can adapt to the variation of environmental factors by adjusting the repetition times of SSR units (Liu et al., 2006; Li et al., 2013).
4.3 Detection of primer polymorphism
With the reduction of sequencing cost and the expansion of sequencing platform throughput, large quantities of microsatellite sequences can be obtained immediately and eff ectively by random sequencing of whole genome using second generation sequencing technology (Kong et al., 2014; Lin et al., 2015; Gao et al., 2017). For example, Gao et al. (2017) successfully screened 60 pairs of microsatellite primers forEpinephelusawoarabased on Illumina genome-wide random sequencing technology from 2.86 G of raw data. Yang et al. (2015) developed 15 polymorphic microsatellite loci inParargyropseditabased on Illumina sequencing technology from 2.80 G of raw data. In this study, we obtained 24 polymorphic microsatellite primers forA.ommaturusbased on Illumina genome-wide random sequencing technology from 6.60 G of raw data.
In this study, 11 polymorphic loci were found to deviate signifi cantly from Hardy-Weinberg equilibrium (P<0.05). Deviation from Hardy-Weinberg equilibrium is a common phenomenon in fi sh populations, which may be caused by inbreeding, non-random sampling, subgroup structure, genetic drift, overfi shing, the Wahlund eff ect, and invalid alleles (Bergh and Getz, 1989; Castric et al., 2002; Lu et al., 2017; Song et al., 2018). The main reason for the null allele of microsatellite is that the mutation of the fl anking sequence of microsatellite results in the abnormal amplifi cation of the gene (Primmer et al., 1995). The generation of null alleles has a great infl uence on the parameters related to population genetic diversity, resulting in “pseudohomozygote” that causes excessive homozygotes and signifi cantly reduces observed heterozygosity and expected heterozygosity (Paetkau et al., 1995). Five loci (BW20, BW34, BW44, BW74, and BW129) showed evidence of null alleles (null alleles frequency>5%) in this study which confi rms the above hypothesis. As a dominant species,A.ommaturusmay suff er from increasing fi shing pressure (Gu et al., 2013; Han et al., 2013). Therefore, null alleles and overfi shing may cause most sites to deviate from Hardy-Weinberg equilibrium.
Biodiversity-rich organisms can better adapt to various environments, and the analysis of genetic diversity is the basis for the conservation of germplasm resources (Lu et al., 2017). The parameters of genetic diversity obtained in this study showed higher genetic diversity as a whole. The polymorphic information content is an important indicator for evaluating the degree of genetic variation of the population. When PIC>0.5, the locus showed a high degree of polymorphism (Botstein et al., 1980). The polymorphism information content ranged from 0.696 to 0.919, all loci were showed highly polymorphic. Allele heterozygosity within population can refl ect the genetic variation level of population (Leberg, 2002). The high proportion of heterozygotes in the population refl ects a stable genetic structure. Generally speaking, expected heterozygosity is more accurate than observed heterozygosity in measuring population genetic diversity (Nei, 1978). When the value of heterozygosity is between 0.5 and 0.8, it can be considered that the population has high diversity (Takezaki and Nei, 1996).The average expected heterozygosity (He) was 0.880, which showed higher genetic diversity. The results show that the markers selected in this study had abundant polymorphism and could be used as eff ective genetic markers for the analysis of genetic diversity and phylogenetic relationships among populations ofA.ommaturus.
Song et al. (2010a, b) carried out some researches on genetic diversity level and population genetic structure ofA.ommaturusin Northwest Pacifi c Ocean by using mitochondrial DNA and AFLP. Mitochondrial DNA markers detected obvious geographic distance isolation patterns, while AFLP markers only detected weak genetic diff erentiation among populations. There is currently no report on population genetics ofA.ommaturusbased on the description of microsatellite markers, and then the new developed 24 polymorphic primer pairs in the present study could be used to analyze the population genetic structure and evaluate the future genetic variation ofA.ommaturus.
5 CONCLUSION
This is the fi rst attempt to analyze the characteristics ofA.ommaturusmicrosatellites using highthroughput sequencing, and the results provided an eff ective basis for our subsequent development of microsatellites primer pairs. The polymorphism primers obtained in this study will provide eff ectively basis for the comparison and analysis of the genetic structure and genetic characteristics ofA.ommaturusin the future.
6 DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article.
7 ACKNOWLEDGMENT
We thank ZHAO Linlin and LI Yuan for the sample collection.
8 AUTHOR DECLARATION
The authors declare that they have no confl ict of interest.
The procedures implemented in this study follow all applicable international, national, and institutional guidelines on the research involved in human participants and/or animals.
Ethical approval was not required for this study because no endangered animal was involved. All handling ofA.ommaturusspecimens was conducted in strict accordance with Animal Care Quality Assurance in China.
References
Arens P, Bugter R, Van’t Westende W, Zollinger R, Stronks J, Vos C, Smulders M J M. 2006. Microsatellite variation and population structure of a recovering tree frog (HylaarboreaL.) metapopulation.ConservationGenetics, 7(6): 825-835.
Bai C C, Liu S F, Zhuang Z M, Yuan Y J, Liu H B, Dai F Q. 2012. Seventeen polymorphic microsatellite markers developed for the Javelin goby,Synechogobiushasta(Gobiidae).GeneticsandMolecularResearch, 11(2): 1 465-1 468.
Barbará T, Palma-Silva C, Paggi G M, Bered F, Fay M F, Lexer C. 2007. Cross-species transfer of nuclear microsatellite markers: potential and limitation.MolecularEcology, 16(18): 3 759-3 767.
Bergh M O, Getz W M. 1989. Stability and harvesting of competing populations with genetic variation in life history strategy.TheoreticalPopulationBiology, 36(1): 77-124.
Botstein D, White R L, Skolnick M, Davis R W. 1980. Construction of a genetic linkage map in man using restriction fragment length polymorphism.AmericanJournalofHumanGenetics, 32(3): 314-331.
Castoe T A, Poole A W, Gu W J, De Koning A P J, Daza J M, Smith E N, Pollock D D. 2010. Rapid identifi cation of thousands of copperhead snake (Agkistrodoncontortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence.MolecularEcologyResources, 10(2): 341-347.
Chen M, Tan Z Y, Zeng G M, Peng J. 2010. Comprehensive analysis of simple sequence repeats in pre-miRNAs.MolecularBiologyandEvolution, 27(10): 2 227-2 232.
Chistiakov D A, Hellemans B, Volckaert F A M. 2006. Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fi sh genetics.Aquaculture, 255(1-4): 1-29.
Duan Y F, Zhang Z, Li J T, Li J, Liu P. 2016. Bioinformatics and microsatellite sequences analysis of EST Sequence in ridge tail shrimpExopalaemoncarinicauda.FisheriesScience, 35(5): 562-567. (in Chinese with English abstract)
Fan H Y, Ji Y P, Zhang S H, Yuan C T, Gao T X. 2005. Research of fi shery biology of the neritic fi shSynechogobiusommaturusin the area of the Huanghe delta.PeriodicalofOceanUniversityofChina, 35(5): 733736. (in Chinese with English abstract)
Feng J, Zu J Q, Zheng Z M, Chen Y C. 2004. Studies on the individual fecundity ofSynechogobiushasta.JournalofZhejiangOceanUniversity(NaturalScience), 23(4): 302-314. (in Chinese with English abstract)
Gao F T, Shao C W, Cui Z K, Wang S P, Wei M, Chen S L, Yang G P. 2017. Development and population genetic diversity analysis of microsatellite markers inEpinephelusawoara.PeriodicalofOceanUniversityofChina, 47(4): 52-57. (in Chinese with English abstract)
Gao S H, Yu H Y, Wu S Y, Wang S, Geng J N, Luo Y F, Hu S N. 2018. Advances of sequencing and assembling technologies for complex genomes.Hereditas(Beijing), 40(11): 944-963. (in Chinese with English abstract)
Gu C P, Gao P, Chen Y J. 2013. DNA barcodingAcanthogobiusommaturusof Zhejiang coast.JournalofZhejiangOceanUniversity(NaturalScience), 32(6): 488-493. (in Chinese with English abstract)
Guo Z Y. 2014. Tandem Repeats Analysis ofYessoscallop Genome. Ocean University of China, Qingdao. (in Chinese with English abstract)
Han D Y, Xue Y, Ji Y P, Xu B D, Liu H, Ma Q Y. 2013. Trophic and spatial niche of fi ve Gobiid fi shes in Jiaozhou Bay.JournalofFisherySciencesofChina, 20(1): 148-156. (in Chinese with English abstract)
Holt R A, Jones S J. 2008. The new paradigm of fl ow cell sequencing.GenomeResearch, 18(6): 839-846.
Kashi Y, King D G. 2006. Simple sequence repeats as advantageous mutators in evolution.TrendsinGenetics, 22(5): 253-259.
Katti M V, Ranjekar P K, Gupta V S. 2001. Diff erential distribution of simple sequence repeats in eukaryotic genome sequences.MolecularBiologyandEvolution, 18(7): 1 161-1 167.
Kong X L, Chen Z Z, Lin L, Li C H, Xu S N, Liu Y. 2014. Polymorphic microsatellite loci isolated from the yellowbelly threadfi n bream,Nemipterusbathybius.GeneticsandMolecularResearch, 13(3): 5 254-5 257.
Leberg P L. 2002. Estimating allelic richness: eff ects of sample size and bottlenecks.MolecularEcology, 11(11): 2 445-2 449.
Li Q. 2004. Advances in studies on microsatellite markers in the Pacifi c abalone.PeriodicalofOceanUniversityofChina, 34(3): 365-370. (in Chinese with English abstract)
Li Y C, Korol A B, Fahima T, Nevo E. 2004. Microsatellites within genes: structure, function, and evolution.MolecularBiologyandEvolution, 21(6): 991-1 007.
Li Z F, Feng Z L, Zhao L H, Shi Y Q, Li C H, Wang L F, Liu Y J, Zhu H Q. 2013. Frequency and distribution of microsatellites in the whole genome ofVerticilliumalbo-atrum.CottonScience, 25(2): 135-141. (in Chinese with English abstract)
Lin L, Zhu L, Liu S F, Su Y Q, Zhung Z M. 2011. Polymorphic microsatellite loci for the Japanese anchovyEngraulisjaponicus(Engraulidae).GeneticsandMolecularResearch, 10(2): 764-768.
Lin L, Li C H, Chen Z Z, Xu S S, Liu Y. 2015. Development and characterization of twenty-three microsatellite markers for the purpleback fl ying squid (Symplectoteuthisoualaniensis).ConservationGeneticsResources, 7(1): 161-163.
Liu L, Li C Y, Yang J, Ye Y F, Li J B, Zhou X G, Wang Y Y, Zhu Y Y. 2006. Analysis of microsatellite sequences in the genome of basidiomycetes fungus,Coprinuscinereus.SouthwestChinaJournalofAgriculturalSciences, 19(1): 131-135. (in Chinese with English abstract)
Lu Y X, Di M Y, Li Y F, Zhou Z C, Hou H M, He C B, Wang S Z, Gao M L. 2017. Microsatellite analysis of genetic diversity in wild and cultured populations of jellyfi shRhopilemaesculentum.FisheriesScience, 36(4): 472-479. (in Chinese with English abstract)
Ma H Y, Chen S L. 2009. Isolation and characterization of 31 polymorphic microsatellite markers in barfi n fl ounder (Veraspermoseri) and the cross-species amplifi cation in spotted halibut (Veraspervariegatus).ConservationGenetics, 5(10): 1 591-1 595.
Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals.Genetics, 89(3): 583-590.
Primmer C R, Møller A P, Ellegren H. 1995. Resolving genetic relationships with microsatellite markers: a parentage testing system for the swallowHirundorustica.MolecularEcology, 4(4): 493-498.
Rousset F. 2008. GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux.MolecularEcologyNotes, 8(1): 103-106.
Sambrook J, Russell D W. 2001. Molecular Cloning. 3rdedn. Cold Spring Harbor Laboratory Press, New York. p.543-554.
Song N, Gao T X, Sun X F, Liu B Z. 2010. Study on species validation forSynechogobiushasta.ActaZootaxonomicaSinica, 35(2): 352-359. (in Chinese with English abstract)
Song N, Zhang X M, Sun X F, Yanagimoto T, Gao T X. 2010a. Population genetic structure and larval dispersal potential of spottedtail gobySynechogobiusommaturusin the North-west Pacifi c.JournalofFishBiology, 77(2): 388-402.
Song N, Zhang X M, Gao T X. 2010b. Genetic diversity and population structure of spottedtail goby (Synechogobiusommaturus) based on AFLP analysis.BiochemicalSystematicsandEcology, 38(6): 1 089-1 095.
Song N, Song L, Gao T X, Sun X F. 2011. Comparative analysis of genetic diversity ofSynechogobiusommaturusbased on the mitochondrial DNA control region.JournalofFisheriesofChina, 35(3): 321-326. (in Chinese with English abstract)
Song N, Li P F, Zhang X M, Gao T X. 2018. Changing phylogeographic pattern ofFenneropenaeuschinensisin the Yellow Sea and Bohai Sea inferred from microsatellite DNA: implications for genetic management.FisheriesResearch, 200: 11-16.
Tόth G, Gáspári Z, Jurka J. 2000. Microsatellites in diff erent eukaryotic genomes: survey and analysis.GenomeResearch, 10(7): 967-981.
Takezaki N, Nei M. 1996. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA.Genetics, 144(1): 389-399.
Uller T, Sagvik J, Olsson M. 2006. Crosses between frog populations reveal genetic divergence in larval life history at short geographical distance.BiologicalJournaloftheLinneanSociety, 89(1): 189-195.
Wang H, Zhang B W, Shi W B, Luo X, Zhou L Z, Han D M, Chang Q. 2012. Structural characteristics of di-nucleotide/tetra-nucleotide repeat microsatellite DNA inPachyhynobiusshangchengensisgenomes and its eff ect on isolation.BiodiversityScience, 20(1): 51-58. (in Chinese with English abstract)
Wang J L, Zhu M X, Xu M H, Chen S L, Zhang F Q. 2017. Analysis on SSR inSinoswertiatetrapterabase on RADseq.BulletinofBotanicalResearch, 37(3): 447-452, 460. (in Chinese with English abstract)
Wang S, Zhang L L, Matz M. 2009. Microsatellite characterization and marker development from public EST and WGS databases in the reef-building coral Acropora millepora (Cnidaria,Anthozoa,Scleractinia).JournalofHeredity, 100(3): 329-337.
Winkelmann I, Campos P F, Strugnell J, Cherel Y, Smith P J, Kubodera T, Allcock L, Kampmann M L, Schroeder H, Guerra A, Norman M, Finn J, Ingrao D, Clarke M, Gilbert M T. 2013. Mitochondrial genome diversity and population structure of the giant squidArchiteuthis: genetics sheds new light on one of the most enigmatic marine species.ProceedingsoftheRoyalSocietyB—BiologicalSciences, 280(1759): 20130273.
Yang B, Lin L, Li C H, Xu S N, Liu Y, Xiao Y Y, Chen Z Z. 2015. Development and evaluation of microsatellite markers inParargyropsedita.SouthChinaFisheriesScience, 11(4): 116-120. (in Chinese with English abstract)
Zane L, Bargelloni L, Patarnello T. 2002. Strategies for microsatellite isolation: a review.MolecularEcology, 11(1): 1-16.
Zerbino D R, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs.GenomeResearch, 2018, 18(5): 821-829.
Zhang D X, Hewitt G M. 2003. Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects.MolecularEcology, 12(3): 563-584.
Zhang G, Fang X D, Guo X M, Li L, Luo R B, Xu F, Yang P C, Zhang L L, Wang X T, Qi H G, Xiong Z Q, Que H Y, Xie Y L, Holland P W H, Paps J, Zhu Y B, Wu F C, Chen Y X, Wang J F, Peng C F, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z Y, Zhu Q H, Feng Y, Mount A, Hedgecock D, Xu Z, Liu Y J, Domazet-Lošo T, Du Y S, Sun X Q, Zhang S D, Liu B H, Cheng P Z, Jiang X T, Li J, Fan D D, Wang W, Fu W J, Wang T, Wang B, Zhang J B, Peng Z Y, Li Y X, Li N, Wang J P, Chen M, He Y, Tan F J, Song X R, Zheng Q, Huang R M, Yang H L, Du X D, Chen L, Yang M, Gaff ney P M, Wang S, Luo L H, She Z C, Ming Y, Huang W, Zhang S, Huang B Y, Zhang Y, Qu T, Ni P X, Miao G Y, Wang J Y, Wang Q, Steinberg C E W, Wang H Y, Li N, Qian L M, Zhang G J, Li Y R, Yang H M, Liu X, Wang J, Yin Y, Wang J. 2012. The oyster genome reveals stress adaptation and complexity of shell formation.Nature, 490(7418): 49-54.
Zhao S L, Wu H L. 2008. Gobiidae:Acanthogobius.In: Wu H L, Zhong J S eds. Fauna Sinica, Ostichthyes, Perciformes (V), Gobioidei. Science Press, Beijing. p.211-215. (in Chinese)
 Journal of Oceanology and Limnology2020年6期
Journal of Oceanology and Limnology2020年6期
- Journal of Oceanology and Limnology的其它文章
- Eff ects of vitamin C defi ciency or excess on growth performance, anti-oxidative response and fatty acid composition of juvenile abalone Haliotis discu s hannai Ino*
- Leptolaimus holovachovi sp. nov. (Nematoda) from Shenzhen Mangrove Nature Reserve, Shenzhen, South China*
- Digging out molecular markers associated with low salinity tolerance of Nannochloropsis oceanica through bulked mutant analysis*
- An enhanced underwater camera apparatus for seabed observation of megabenthic epifauna in the northern Yellow Sea*
- Characteristics of zooplankton community in North Yellow Sea unveiled an indicator species for the Yellow Sea Warm Current in winter: Euchaeta plana*
- Intraguild predation by polyps of three scyphozoan jellyfi sh: Nemopilema nomurai, Aurelia coerulea, and Rhopilema esculentum*