
气相色谱-燃烧-同位素比质谱在检测尿样中外源性睾酮及其代谢物中的应用
2020-11-24 08:48:36温超王静竹朱天硕刘欣王杉张亦农
中国运动医学杂志 2020年9期
温超 王静竹 朱天硕 刘欣 王杉 张亦农
国家体育总局反兴奋剂中心(北京100029)
1 引言
睾酮(testosterone,T)是人体内主要的固醇激素之一,可以促进机体的合成代谢,促进肌肉和力量的增长[1-3],因此被某些运动员作为兴奋剂使用。由于体内自身合成的睾酮和睾酮制剂中的睾酮的分子结构完全一致,常规的质谱仪难以区分体内自身合成的睾酮和外源摄入的睾酮。同时,由于个体差异、不同的生理状况,尿样中的睾酮浓度变化范围较大,因此,尿样中外源性的睾酮确证,一直以来都是兴奋剂检测领域的重点和难点[4-6]。按照世界反兴奋剂机构(WADA)的规定[7,8],当体内睾酮、表睾酮及其代谢物的浓度或者比例发生异常变化时,如睾酮和表睾酮浓度比例大于4,或者在男性尿样中睾酮含量大于200 ng/mL 时,需要对尿样中的睾酮以及其主要代谢物进行气相色谱-燃烧-同位素比质谱(GC-C-IRMS)确证[7-12],区分被分析物是否为外源性类固醇激素。
GC-C-IRMS 检测技术在兴奋剂检测领域已经有20年左右的应用[13-21]。通过对样品待测物中13C/12C 的比例与内源性激素参照物中13C/12C 比例进行比较,可以判断样品中待测物是否为内源产生或外源摄入。通常13C/12C 的比例表示为δ13C 值,使用维也纳Pee Dee 箭石标准(Vienna Pee Dee Belemnite,VPDB)作为国际单位进行校准[22]。
目前,大多数人工合成的类固醇激素均来自于植物基质,相对于人体自身合成的类固醇激素,人工合成的类固醇激素具有更贫乏的δ13C值[7,17,22-24]。当人体摄入外源性睾酮,其体内睾酮及其代谢物的δ13C值将随之变化,而与睾酮代谢无关的其他类固醇激素则不受影响。在兴奋剂尿样检测中,常采用指定一种内源性类固醇激素为内源性参照化合物(endogenous reference compound,ERC),将其的δ13C值与待测检测目标化合物(target compound,TC)的δ13C 值进行比较,可以判断出TC 是否与ERC 一样为内源性产生。在兴奋剂检测领域,孕烷二醇(pregnanediol,PD)、11β-羟基-雄酮(11βhydroxyandrosterone,11OHAn)、5α-雄烷-16-烯-3β-醇(5α-androst-16-en-3β-ol,16en)、11-ketoetiochola⁃nolone(11-keto)等常被作为内源性参照化合物[7]。按WADA 技术文件规定,当ERC 和TC 的δ13C 值差(表示为Δ)大于3‰或4‰时(根据TC 种类而定),则认为该TC来自于外源性摄入。
当样品进入GC-C-IRMS 后,先经过气化,然后在气相色谱中进行分离,分离后的组分经过燃烧管转化为CO2后再进入同位素比质谱进行检测,最终得到待测物质的δ13C 值。由于GC-C-IRMS 分析仅能对待测物的δ13C值进行测定,无法对待测样品的结构信息进行确认,因此当待测物质在气相色谱有干扰或者共流出峰时,测定的δ13C值将不准,无法用于结果判断。所以,样品的前处理对于GC-C-IRMS 分析极其重要。由于尿样中基质成分复杂,而待测物睾酮及其代谢物浓度较低,如何采用高效的纯化分离技术对尿样中的待测物质进行分离提纯,并测定其δ13C值,是兴奋剂检测领域中亟待解决的技术之一。目前国际常用的方法是使用两次液相分离纯化,提高分析物的纯度,或通过化学衍生化手段,使分析物在气相色谱上完全分离[9-12,23-24],这些方法可以有效提高尿样中待测组分的分离效率,但需要大量时间和人力进行监测分析。
目前国际通用的方法大多基于Piper 等的两次液相色谱纯化结合衍生化法[17]。该方法先将尿样进行酶解处理,然后进行第一次液相色谱分离,通过收集经过液相洗脱的待测物质馏分达到分离纯化作用。在经过第一次液相分离后,将其中难以分离的待测组分,睾酮、5α-雄烷-3α,17β-二醇、5β-雄烷-3α,17β-二醇物质馏分进行乙酰化反应,将待测物质转化为相应的乙酰化产物。对经过乙酰化的组分进行第二次液相色谱分离,收集对应的馏分。经过以上液相色谱分离的样品可以进行GC-C-IRMS 分析。通过两次液相分离以及衍生化反应,可以对尿样中的睾酮及其代谢物进行纯化,满足GC-C-IRMS测试的样品需求。该方法的两次液相色谱分离以及衍生化反应需要大量时间以及人工操作,在大型体育比赛及样品高峰期间,会对常规检测工作带来巨大压力。同时,由于对待测物质进行衍生化后化学基团发生转化,因此测定的δ13C值需要进行校正,该过程也会产生一定测量误差。
建立高效快捷的GC-C-IRMS检测方法,对于我国兴奋剂检测工作具有重要现实意义。本研究根据WA⁃DA的相关技术文件要求进行实验方案设计,建立检测方法,主要对方法的稳定性、重现性以及线性、不确定度、人群值等进行验证评价,建立一套GC-C-IRMS 检测方法,用于对尿样中睾酮及其代谢物进行来源分析。
2 实验部分
2.1 仪器与试剂
E.coli IX-A 型β-葡萄糖醛酸甙酶购置于Sigma公司;睾酮、表睾酮(epitestosterone,ET)、孕二醇、5α-雄烷-3α,17β-二醇(5α-androst-3α,17β-diol,5α-di⁃ol)、5β-雄烷-3α,17β-二醇(5β-androst -3α,17β-di⁃ol,5β-diol)、雄酮(androsterone,An)、本胆烷醇酮(etiocholanolone,Etio)、11-β-羟基雄酮、甲基睾酮(17α-Methyltestosterone,MT)标准物质均购置于澳大利亚国家计量研究所;磷酸二氢钾(KH2PO3)、磷酸氢二钾(K2HPO3)、乙酸乙酯、正己烷等购置于国药集团化学试剂有限公司;甲醇、正己烷(C6H14)、乙酸乙酯(C4H8O2)、乙腈(CH3CN)、叔丁基甲醚((CH3)3COCH3)购置于Sigma-Aldrich 公司;所用的氦气、二氧化碳以及氧气来自于北京氦普北分气体工业有限公司。
液相色谱采用安捷伦公司Agilent 1200 series HPLC,配有Agilent 1260 Infinity collector 系列接收器,气相色谱-燃烧-同位素比质谱来自于赛默飞世尔科技有限公司,配置有Trace1310 系列气相色谱仪Thermo Scientific MAT 253同位素比质谱。
2.2 本研究的实验原理和方法
2.2.1 实验原理
待测物睾酮及其代谢产物在尿样中均以葡萄糖苷酸形式存在,此类结合形式由于沸点高等原因,无法使用气相色谱进行分离,因此无法进行GC-C-IRMS 分析。在实际尿样检测中,使用β-葡萄糖醛酸甙酶将尿样中的待测物质进行酶解,将其转化为游离态化合物后再进行GC-C-IRMS分析[7]。
GC-C-IRMS 检测原理为将待测物质通过气相色谱气化分离后,经由燃烧管转化为CO2,通过分析CO2中的13C/12C 的比例计算得到δ13C 值,即为待测物质的δ13C值。当待测物质中有干扰物时,会在气相色谱中出现共流出峰,测定出的δ13C值为待测物和干扰物的混合值,无法进行结果判定。因此,在GC-C-IRMS 检测过程中,样品的前处理非常重要。虽然气相色谱具有一定分离能力,但由于尿样基质复杂,气相色谱的分离能力已经无法满足实验要求。尿样中基质种类复杂,因此需要在GC-C-IRMS 分析前将尿样中的待测组分进行分离富集,得到纯度较高的样品。在实际尿样检测中,通常使用液相色谱将尿样中的待测物质进行分离纯化,其原理为将尿样中的待测组分在液相色谱柱上进行洗脱[9],按照标准物质的保留时间,收集纯化后的液相色谱馏分[6,7,9,17]。该馏分中含有纯度较高的待测物质,后续经过气相色谱分离,可以得到纯净的气相色谱峰,对待测物质进行δ13C值测定。
2.2.2 实验方法
现将实际检测中的样品处理操作举例说明。取适量尿样,以待测物浓度100 ng/mL 的尿样为例。取2份,每份3 mL尿样,每份加入1 mL磷酸缓冲液(pH约为6.0~7.0),加入100μL β-葡萄糖醛酸甙酶(5000 units),55℃恒温水浴保温180分钟以上或过夜,取出样品冷却至室温,加入4 mL叔丁基甲基醚,振荡萃取,离心后取上层有机相,75℃加热下氮气吹干,冷却至室温,加入65 μL含有内标MT浓度为的0.3 mg/mL的甲醇溶液,振摇后转移至液相进样瓶中,封口。经过离心后静置待用。样品采用HPLC 分离纯化。选用色谱柱ZORBAX SB-C18(4.6 μm×250 μm,5 μm),梯度如下:80∶20(A∶B)→15 min→50∶50(A∶B)-保持10min→8 min→10∶90(A∶B)→1min→0∶100(A∶B)-保持5min→4 min→80∶20(A∶B),平衡6 min.流速1.0 mL/min,柱温35 ℃。流动相A 为水,B为乙腈。通过检测在192和244 nm处的紫外检测信号来对样品收集时间以及运行稳定性进行判断。HPLC分离组分经过收集后,在75℃加热下氮气吹干待用。使用20 μL IRMS 上机溶液进行溶解封口后进行GCC-IRMS分析。
3 结果与分析
本研究根据WADA技术文件进行实验设计和方法验证,以达到WADA对于GC-C-IRMS检测的技术要求和相应指标。本方法中所有测定的δ13C值均使用可溯源标准品CU/PCC34-3 对仪器参考气进行校准。本方法涵盖7种物质的检测,其中PD和11OHAn为ERC,T、5α-雄烷-3α,17β-二醇(5α-diol)、5β-雄烷-3α,17β-二醇(5β-diol)、雄酮(An)、本胆烷醇酮(Etio)为TC。在实际样品中,T、5α-diol、5β-diol 三种物质的浓度往往低于其他的物质。
3.1 液相色谱分离
为进一步提升待测物质ERC 和TC 的纯度以及对样品进一步富集,需要在进行GC-C-IRMS 检测前,对尿样中的待测物质进行液相色谱分离提纯,收集经液相色谱柱分离后的各个组分的流出物,该过程可以将尿样中的大量基质排除,有效提高待测物质的纯度。尿样通过液相色谱柱的过程中,会产生同位素分馏效应[25-26],流出物的收集时间差异导致最终测定结果的δ13C 值出现偏差。根据WADA 技术文件要求,当Δ大于3‰或4‰时(根据TC种类而定)时可判定待测物TC为外源摄入,因此在液相色谱纯化过程中一定要避免同位素分馏效应对最终结果的影响,造成假阳性或假阴性。
在方法验证过程中,须将标准物质在液相分离前后进行对比,确认收集时间窗口内收集的样品未受到同位素分流效应影响。图1为标准物质经过液相纯化的色谱图,由图中可见,所有的待测物质,包括ERC 和TC,均可以较好分离。通过测定标准品经过液相色谱分离前后的δ13C值的差异,可以对收集时间窗口进行评估。经过优化后,各物质在液相色谱出峰前0.05 min和出峰后0.12 min进行收集为最佳的液相色谱收集窗口。图2为一份真实尿样的液相色谱分离谱图。实验中使用9 mL 尿样进行测定,由图可知,由于尿样中存在大量基质,液相色谱谱图极为复杂,无法对样品中各组分进行区分。因此前期的收集窗口确认对样品的纯化分离有重要影响。在实际样品运行序列中,需要在序列前后分别加入标准物质对收集窗口的有效性进行确认。同时,通过使用具有紫外吸收的内标物质甲睾(MT)作为内标,监测样品中MT保留时间的变化,可以对样品液相色谱运行情况进行评估。收集后的待测物质重新定容进行GC-C-IRMS 分析。通过真实尿样的分析物浓度及其最终在GC-C-IRMS 上的信号相应进行折算,测定本方法液相分离过程的回收率在70%左右。

图1 各分析物的标准品的液相色谱图及时间收集窗口
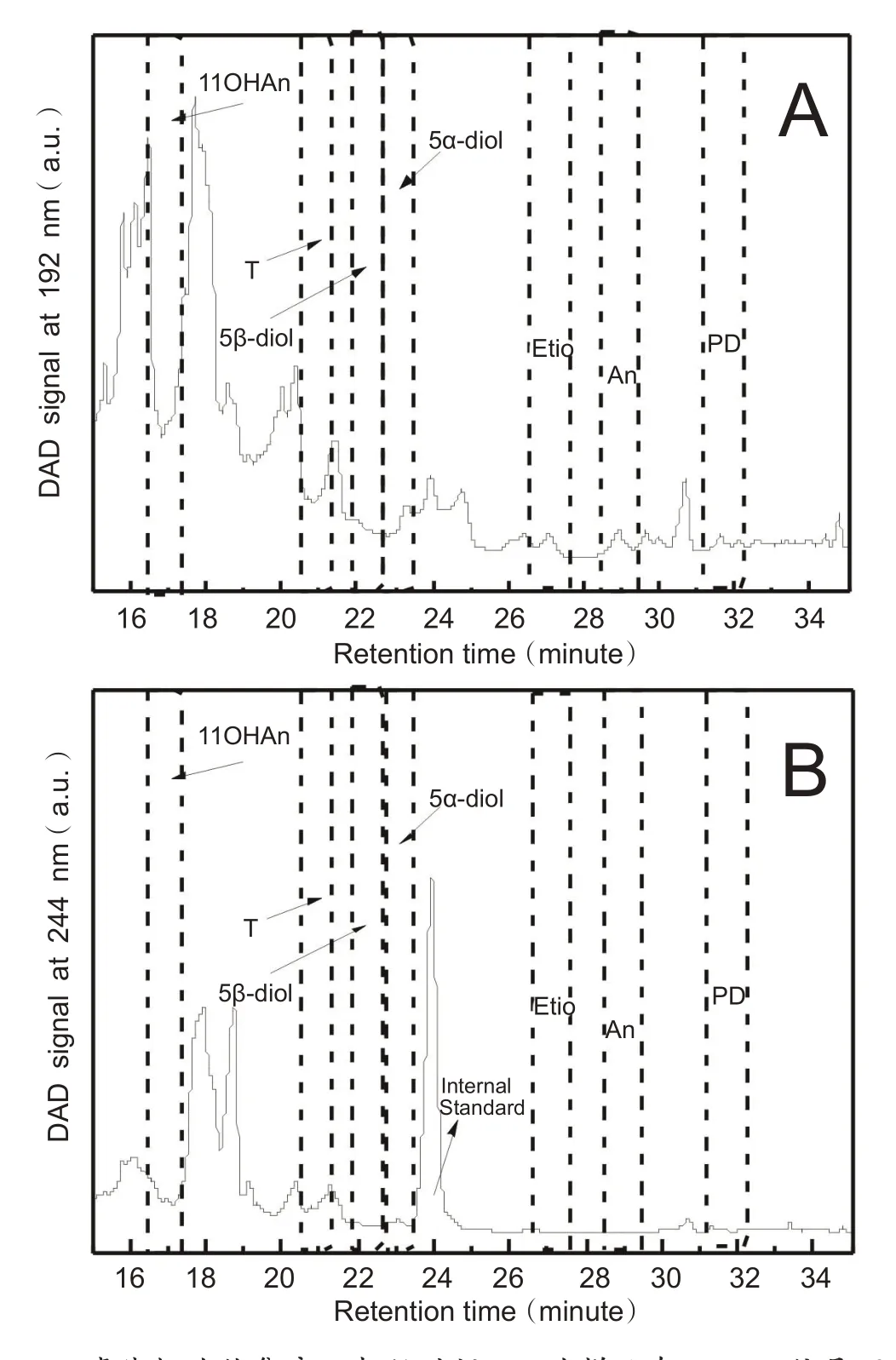
图2 真实样品液相色谱图
3.2 液相色谱稳定性评价
液相色谱的保留时间稳定性对样品中的待测物质检测具有较大影响,本实验通过标准物质保留时间窗口确认,应用于尿样中待测组分进行分离富集,因此保留时间是否稳定对最终检测结果至关重要。在尿样检测过程中,要求尿样中内标物质的保留时间与该批次标准物质中内标在液相纯化过程中的保留时间绝对差值小于0.1分钟,以避免在待测物收集过程中产生的同位素分馏效应[26]。本研究提供的检测方法具有较高稳定性,在液相色谱柱以及液相色谱主要备件不更换的状态下,液相色谱柱前压波动小于10 bar,各批次中尿样中内标物之保留时间偏离标准物质保留时间超过0.1分钟占总样品约为3%。考虑到不同尿样中基质不同,因此在实际检测中出现保留时间偏差是不可避免的。当实际样品运行中内标保留时间与该批次标准物质运行保留时间差绝对值超过0.1分钟时,应对该样品进行重新分析,并在收集时适当扩大收集窗口以保证待测物质可以完全收集。
3.3 仪器线性范围测定
GC-C-IRMS 的工作原理为待测物质经过GC分离后通过燃烧管转化为CO2,再对产生的CO2进行δ13C 进行测定,即可得到待测样品的δ13C值。在样品经进样口进入气相色谱过程中,由于不同进样条件、进样量,进样浓度等都会发生同位素分馏现象[26],导致测定的δ13C发生偏差,因此需要对仪器的线性条件进行评价。按照WADA技术文件要求,GC-C-IRMS方法的仪器线性评价至少要包含8 个以上进样量,同时要求每种物质测定的δ13C 值的标准偏差(SD)须小于0.5。在本研究方法中,我们将每个标准品配制成三份不同浓度的溶液,每份溶液使用0.5、1.0、1.5、2.0、2.5、3.0 μL 进样测试,得到一系列每种分析物在不同信号下的δ13C值。表1列出了使用标准物质对仪器线性的测定结果。根据所得到的测量结果可知,所有的检测物质在300~5000 mV 具有稳定的δ13C 值。因此,在实际检测中,需要对处理后的样品在上机前进行进一步稀释,得到合适的浓度,在1~2.5 μL的进样条件下样品信号在仪器线性范围内才能得到准确的值。

表1 GC-C-IRMS仪器线性范围测定
3.4 检测浓度线性范围
由于个体差异及环境差异,不同尿样中的TC 和ERC 浓度会有极大差别[7,12],因此GC-C-IRMS 确证方法测定的δ13C 值不能受到尿样中待测物质的浓度的影响。按照WADA 技术文件规定,对于同一来源的待测物在不同浓度下(例如同一人在不同时间段留取的尿样中待测物质浓度不同)的δ13C值测定结果应一致。该评价又称为方法的检测浓度线性范围。在本方法评价过程中,选取的尿样检测浓度可以覆盖当前我国兴奋剂检测实验室的常规检测中的待测物尿样浓度范围。如表2所示,同一待测物质在不同的浓度下测定的δ13C值非常稳定,测定的SD均小于0.5,表明本研究的方法可以对不同浓度的尿样进行检测。

表2 GC-C-IRMS的检测浓度线性范围
3.5 最低检测浓度
由于GC-C-IRMS的灵敏度所限,通常需要至少15纯物质进入气相色谱柱才能得到可靠的信号,因此在常规检测中,对于低浓度的尿样,需要提高样品使用量来得到稳定的信号。根据WADA 要求,每份尿样中的A瓶尿量不小于45 mL,B瓶尿样量不小于30 mL。兴奋剂检测常为多个方法对不同类别兴奋剂药物同时检测,对可疑样品须进行确证程序,因此各检测方法取用尿样应尽量少,以保证其他检测方法具有足够检测尿样可用。本研究方法中最低检测浓度是基于15 mL尿样而测定的最小待测物质浓度。通常An 和Etio 在尿样中浓度较高,因此本方法仅验证了100 ng/mL 尿样。其他的ERC 验证了10 ng/mL 尿样。在进行验证过程中,分别连续三天进行三次测定,同一分析物得到三份结果。从表3的比对结果中可知,在采用15 mL尿样分析时,使用本方法可以对含量为10 ng/mL 的PD、11OHAn、T、5α-diol、5β-diol测定δ13C值,且结果稳定。

表3 GC-C-IRMS方法最低检测浓度测定
3.6 不确定度
本文中方法的结合不确定度包括方法中间精密度和偏差的均方根。具体公式如下:
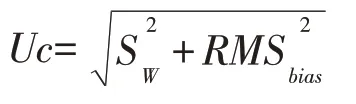
其中Uc为方法的不确定度,Sw为方法的中间精密度的贡献,RMSbias为方法测量偏差贡献。实际测定中Sw为10 份阴性质控尿样和10 份阳性质控尿样的测量结果的标准偏差。而方法测量偏差是通过Keeling plot进行线性拟合结果得到[7,12,17]。表4列出了不确定度的评价结果,从表中可知,所测的质控样品Sw分别为0.5~0.7 之间,表明本方法具有较高稳定性,所有测定的不确定度均小于1‰,满足WADA 对于兴奋剂检测的要求。
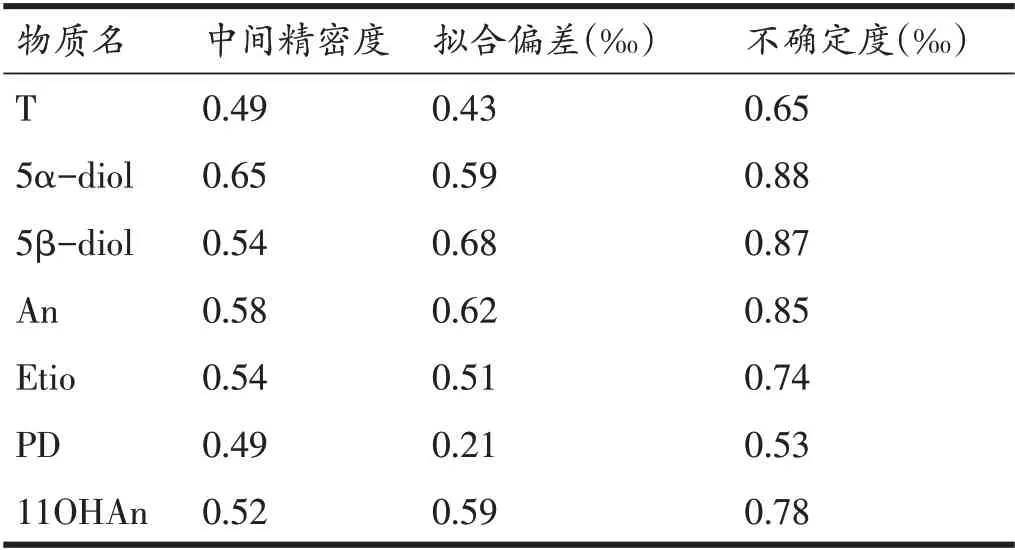
表4 GC-C-IRMS方法不确定度测定
3.7 人群值分析
通过GC-C-IRMS方法比较尿样中的ERC与TC的δ13C值差异,可以判断TC是否与ERC一样为内源性代谢产生。按照WADA 技术文件要求,当ERC 和TC 的Δ>3‰或4‰时,可以判定测定的TC 为外源性摄入。在实际检测中,需要对方法的检测的人群值进行评价,通过测定40份阴性样本,得到样本的ERC和TC的Δ值和对应Δ值的SD,要求ERC 与TC 的平均Δ±2SD≤3‰。表5列出了本研究方法的人群值分析结果。如表中所示,所有的分析结果的Δ±2SD 值均小于3‰,满足WA⁃DA要求,表明本方法具有极高稳定性。

表5 GC-C-IRMS方法人群值分析
4 结论
本研究建立了一套完整的GC-C-IRMS 方法对尿样中的睾酮及其主要代谢物的来源进行区分。尿样经液相色谱纯化分离,进行富集,有效提高了分析物的纯度,通过测定,样品前处理回收率在70%以上。本方法在取用15 mL 尿样的情况下,可以对10 ng/mL 的睾酮及其主要代谢产物进行GC-C-IRMS 检测。不同浓度待测物的尿样检测结果,以及不确定度、人群值分析结果表明本方法测定结果较为稳定,满足WADA 对于兴奋剂检测的要求,可以适用于兴奋剂检测实验室常规检测以及大赛检测。
猜你喜欢
现代泌尿生殖肿瘤杂志(2022年1期)2022-11-21 14:37:55
家庭科学·新健康(2022年2期)2022-03-07 06:13:38
成都体育学院学报(2021年1期)2021-07-16 07:37:32
中国体育教练员(2017年3期)2018-01-19 02:41:52
中国体育教练员(2017年2期)2017-07-31 19:09:34
发明与创新(2016年33期)2016-08-21 13:22:20
大众健康(2016年3期)2016-05-31 23:59:46
环球时报(2016-05-18)2016-05-18 08:49:41
色谱(2015年6期)2015-12-26 01:57:30
中国环境科学(2015年7期)2015-01-28 11:41:53
