
交联质谱技术研究进展
2020-11-23 12:04刘春丽韩碧莹赵跃芳
化学与生物工程 2020年11期
张 洁,刘春丽,李 欣,张 媛,韩碧莹,赵跃芳
(内蒙古大学 省部共建草原家畜生殖调控与繁育国家重点实验室,内蒙古 呼和浩特 010070)
蛋白质作为生命功能的“执行者”,在生命体中通过高度复杂且动态变化的相互作用网络调控各种生物过程,蛋白质相互作用的研究是阐明生物学过程的关键。传统蛋白质相互作用的研究方法主要有酵母双杂交筛选(Y2H)法、免疫共沉淀(Co-IP)法、亲和纯化与质谱联用(AP-MS)法[1]、邻近依赖性生物素鉴定(BioID)法[2]等,这些方法虽然已经应用到许多生物学研究中,但具体操作仍存在一定的局限性。Y2H法假阳性较高,由于融合蛋白受表达条件限制,可能导致蛋白质的修饰与折叠发生改变,不能形成正确的相互作用关系; Co-IP法与AP-MS法灵敏度较低,无法捕捉瞬时与低亲和力的相互作用蛋白,且不能区分作用方式[3];BioID法可用于蛋白质相互作用的筛选,但由于需要添加过量生物素,可能对细胞产生毒害作用,而且生物素化会改变蛋白的电荷与修饰,影响邻近蛋白的行为。
交联质谱技术(XL-MS)是将蛋白质化学交联技术与质谱技术相结合,用于研究蛋白质结构以及蛋白质相互作用的新方法。通过在样品中加入适量的小分子化学交联剂,使空间足够接近的氨基酸与交联剂发生共价连接,从而将已经发生的相互作用稳定下来,通过质谱与交联质谱数据处理软件解析确定发生相互作用的蛋白质以及具体作用的交联位点,获取蛋白质相互作用信息,结合交联剂的长度对蛋白质的空间结构及蛋白质复合体亚基的空间排列进行预判[1]。
XL-MS的优势[4]在于:(1)样品制备简单。蛋白质发生交联后经过常规蛋白质组学的酶切、纯化、质谱分析便可得到交联信息,操作简便,不受分子量以及样品纯度限制;(2)检测通量高。基于质谱技术特点,可以高通量地同时进行多种蛋白质的结构和相互作用的分析;(3)灵敏度高。生物反应转瞬即逝,交联剂可以使瞬时相互作用和结合不牢固的反应的特定氨基酸发生共价连接,揭示瞬时或者微弱的蛋白质相互作用;(4)作用方式明确。XL-MS可确定蛋白质之间是直接相互作用还是间接相互作用,结合晶体结构信息可以明确相互作用界面信息;(5)应用范围广。交联剂除了可以在蛋白分子水平上交联,部分交联剂还可进入细胞内交联,在细胞水平上实现生理状态的相互作用研究。
1 交联质谱技术的工作流程
XL-MS的工作流程(图1)包括:蛋白质交联反应的进行(体外纯化蛋白质复合物交联、亲和纯化蛋白质交联、体内交联);交联蛋白质或肽段的富集纯化;交联蛋白质的酶切;交联多肽的质谱鉴定;质谱数据的解析与交联位点的确定,结合已有的蛋白质结构信息进行复合体结构模型建立,蛋白质相互作用网络分析和亚基拓扑图谱分析。

图1 XL-MS的工作流程
蛋白质交联反应是XL-MS分析的关键步骤之一,直接影响着实验结果的成败。在研究简单蛋白质或蛋白质复合体亚基的相互作用界面时,要预先根据目的蛋白的结构及感兴趣的结构域选择合适的交联剂,在不影响蛋白质正常构象且交联剂反应活性较高的条件下进行交联,通过确定交联剂与目的蛋白反应比例、反应酸碱度与反应时间等,使交联剂与蛋白质和缓冲液体系相匹配,避免破坏蛋白质天然结构而引起交联反应过度或不足,出现少交联或假交联的错误信息[4];在研究未知蛋白质相互作用时,交联剂中间臂的长度也是重要影响因素。
酶切反应通常选用常规的胰蛋白酶(trpsin)切割精氨酸(Arg)和赖氨酸(Lys)的C端。Leitner等[5]研究发现,除了使用胰蛋白酶外,添加4种蛋白酶天冬氨酸(Asp)-N、谷氨酸(Glu)-C、Lys-C和Lys-N等多酶酶切可以增加交联位点的鉴定数。
交联肽段的特异性富集和分离,可以提高交联肽段的信号强度并降低样品的复杂性,提高交联肽段的鉴定效率,根据富集时间的不同分为消化前蛋白质富集和消化后肽段富集。消化前蛋白质富集指对感兴趣的细胞器或目标蛋白质进行提纯或靶向富集,即将交联与亲和纯化相结合,通过亲和纯化钓取交联反应后目标蛋白质复合物或先钓取目标蛋白质复合物再进行交联反应,可以有效降低交联肽段鉴定的复杂性[6]。交联蛋白也可通过SDS-PAGE胶内分离与酶解。在质谱数据采集时,可以通过设定二级质谱条件采集,只采集高价态离子的二级质谱信息,提高交联肽段的鉴定率[7]。消化后肽段富集指将发生交联的肽段与非交联的线性肽进行分离,常用的方法是尺寸排阻色谱法(size exclusion chromatography,SEC)。Leitner等[5]使用SEC分析,基于其较高的分子量选择交联线性肽段,从而减少了存在于交联体系中大多数非交联的线性肽,证明SEC是一种有效的交联肽段富集方法。此外,由于交联肽段的电荷数增加,交联肽段比线性肽拥有更多的带正电荷的碱性氨基酸,可使用强阳离子交换色谱法(strong cation exchange chromatography,SCX)进行富集分离[8]。
2 交联质谱技术的主要应用
2.1 蛋白质结构的研究
蛋白质结构研究的常规方法有:X-射线晶体衍射法、核磁共振波谱法等。虽然这些方法具有精密度高、获得信息丰富等优点,但对于蛋白质样品的纯度、浓度、分子大小及可结晶性等要求很苛刻,不利于对大分子蛋白质复合体结构的解析。而XL-MS在这一方面起到不可或缺的作用,当电镜的分辨率不够或蛋白难以形成结晶时,可通过XL-MS结果解析出蛋白质不同结构域或不同亚基之间的相互作用界面[9]。通过软件预测出多种可能构象后,XL-MS数据可以辅助排除一些不合理的构象[10]。XL-MS以其独特优势,与低温冷冻电子显微镜(cryo-EM)、X-射线晶体衍射法等其它研究蛋白质结构的方法相结合,在超大分子量蛋白质复合体结构的解析中起到了提供距离约束、指导计算模型确定亚结构作用位点的重要作用,体现了其在蛋白质结构研究中的实用性和重要性[1]。
核糖体蛋白S1由于其动态的特征,无法通过X-射线晶体衍射技术进行观察。2011年,Lauber等[11]通过新型的赖氨酸反应性交联剂(亚乙基次硫亚氨酸二乙酯,DEST),利用XL-MS探测柔性组件中亚基间作用关系的特性,成功定位了大肠杆菌核糖体复合物上核糖体蛋白S1的结合位点[12]。蛋白酶体26S由调控亚基19S和催化亚基20S组成,20S的结构早在1995年通过X-射线晶体衍射技术完成了解析[13],但完整的26S复合体由于其溶解性差、结晶困难未能获得结构信息。2012年,Lasker等[14]通过XL-MS、EM及其它方法的联合使用获得了蛋白酶体26S的完整结构。2014年,Erzberger等[15]将XL-MS、EM、X-射线晶体衍射技术以及综合建模相结合,阐明了酿酒酵母的40S核糖体·eIF1·eIF3复合物的结构,共鉴定到965对蛋白内和蛋白间的交联。线粒体的核糖体在结构和细胞功能上与胞质核糖体差异很大,主要负责合成一定数量的疏水膜蛋白,晶体培养难度大限制了X-射线晶体衍射技术对其结构的解析,在高分辨率cryo-EM研究线粒体核糖体结构的研究中,XL-MS对复合体中单个亚基的定位起到了关键作用[16]。2015年,Martinez-Rucobo等[17]通过XL-MS成功解析了体外重组的加帽酶(capping enzyme,CE)与磷酸化的RNA聚合酶Ⅱ(PolⅡp)形成的转录复合体PolⅡp-CE的结构,共获得527个交联肽段,其中337个符合赖氨酸-赖氨酸交联的距离限制。此项研究再次证实了XL-MS是研究动态柔性蛋白质复合体的有效方法。此外,XL-MS还成功地用于解析TriC/CCT伴侣蛋白系统[18]、核孔复合物[19]、CRISPR-Cas复合物[20]以及PolⅡ-介体起始复合物[21]的结构。
2.2 蛋白质相互作用的鉴定
由于AP-MS法与Co-IP法研究蛋白质相互作用存在局限性,可将其与XL-MS结合,在亲和纯化或免疫共沉淀之前进行共价交联,就可以顺利将瞬时或微弱的蛋白质相互作用固定下来,并且通过交联位点的解析获得相互作用的空间界面信息。2012年,Herzog等[22]通过对蛋白质磷酸酶2A(PP2A)信号通路检测到的已知蛋白质进行亲和标记纯化后在磁珠上进行交联,经质谱分析鉴定到涉及特定三聚体PP2A复合物与许多衔接子蛋白之间相互作用的176个蛋白间和570个蛋白内交联,并阐明了免疫球蛋白结合蛋白1(IGBP1)和PP2A之间的结合界面,揭示了TCP1环复合物(TRiC)伴侣蛋白与PP2A调节亚基2ABG相互作用的拓扑结构。2015年,Shi等[23]利用免疫共沉淀结合XL-MS,使用工程设计的GFP纳米抗体从细胞中亲和捕获GFP标记的蛋白质复合物,然后直接在磁珠上进行交联反应,将交联结果结合计算机模拟解析了多个蛋白质复合物的天然结构。
除此之外,XL-MS也可以单独应用于细胞环境(活细胞或细胞裂解液)中研究生理状态下蛋白质的相互作用,捕获生理条件下广谱的瞬时、微弱的蛋白质相互作用。目前XL-MS已经用于几种不同生物全蛋白质组范畴的相互作用研究。2012年,Yang等[24]通过在大肠杆菌和线虫的裂解液中进行交联,使用pLink软件分别鉴定到394对和39对交联肽对,之后开发了一种类似BS3的以赖氨酸为靶标的可富集的交联剂,在大肠杆菌裂解液中得到3 130个相互作用的赖氨酸肽对,属于677个蛋白质;在线虫裂解液中鉴定到893个相互作用赖氨酸肽对,属于121个蛋白质[25]。2015年,Liu等[26]在HeLa细胞裂解液中利用二琥珀酰亚胺基亚砜(DSSO)交联后鉴定到2 179个独特的赖氨酸-赖氨酸交联肽段,并阐述了80S核糖体核心复合体与纤溶酶原激活物抑制剂 RNA结合蛋白1(SERBP1)之间的相互作用结构细节;2017年,他们在大肠杆菌和HeLa细胞裂解液中进行全蛋白质组范围的相互作用研究,对质谱碎裂方法以及数据分析方法进行优化后,分别鉴定到1 158对和3 301对交联肽对,并重点描述了涉及翻译、蛋白质折叠和碳水化合物代谢的几种蛋白质复合物的结构信息,这些蛋白质相互作用网络为许多内源性大分子复合体结构及相互作用研究提供了重要信息[27]。2018年,Fasci等[28]利用XL-MS绘制了人源细胞核中的蛋白质相互作用图,在确定的8 700个交联肽段的2/3代表不同蛋白质间的相互作用,并重点阐述了核小体上核心组蛋白的相互作用,建立了USF3和Ran GTPase与核小体结合模式的低分辨率模型。诸多的研究证实XL-MS在复杂样品中有着较好的应用,设法在体内(细胞内)进行交联可以在蛋白质复合物保持天然构象且不会破坏翻译后修饰的生理条件下研究蛋白质相互作用。
3 交联质谱技术应用的主要挑战
交联剂、交联肽段的富集以及反应位点具有特异性、交联肽段丰度低、交联肽段谱图复杂、解析困难等诸多因素限制了XL-MS的发展。XL-MS中应用最广泛的交联剂是基于N-羟基琥珀酰亚胺酯类(NHS)的胺基交联剂,但NHS易发生水解,而且当蛋白质缺乏赖氨酸(如膜蛋白的跨膜区域赖氨酸较少)时,胺基交联剂则无法捕捉相互作用蛋白。虽然研究开发了许多靶向其它氨基酸的新型交联剂,但在具体应用时仍会受到一定限制。此外,全蛋白质组范畴的蛋白质交联质谱研究对于交联肽段的富集纯化以及交联肽段谱图的解析提出了更大的挑战,交联蛋白浓度通常很低,有效的富集分离是交联质谱鉴定的关键。而产生的交联肽段极大地增加了谱图解析的复杂度,目前可用的软件工具中,都有其特殊性及一定的适用范围,还没有十分理想的通用软件,交联位点鉴定的可靠性与覆盖率仍需不断提升。
3.1 交联剂
为捕捉与稳定蛋白质间的相互作用可利用反应活性较高的侧链基团,如赖氨酸上的氨基、天冬氨酸和谷氨酸上的羧基、精氨酸上的胍基、半胱氨酸上的巯基,分别设计开发氨基反应的交联剂、羧基反应的交联剂、胍基反应的交联剂以及巯基反应的交联剂,同时可以将这些化学交联剂不断优化,使交联剂具有可进行化学切割、质谱切割或可富集、含标记等不同特性。各种类型的交联剂简单介绍如下:
(1) 氨基反应的交联剂。目前最常用的氨基反应交联剂是NHS与亚氨酸酯。根据实验不同,可选择短臂或长臂、可解离或不可解离、膜通透型或细胞表面型等多种间隔臂类型的NHS,在弱碱性条件下能够与伯胺反应生成酰胺键,具有较高的反应效率,但反应导致的电荷变化可能引起蛋白质构象变化;亚氨酸酯与NHS不同的是它与氨基反应生成碱性脒基,保持了反应位点的正电荷性质,虽然反应效率略低,但特异性高于NHS,水溶性更好。
(2)羧基反应的交联剂。设计开发蛋白质酸性氨基酸的羧基反应基团的交联剂能够扩大XL-MS的应用范围,与羧基反应的常见化学基团主要有碳化二亚胺(EDC)、重氮烷和羰二咪唑。EDC在酸性条件下具有较高的反应活性,ADH和PDH能够实现生理条件(pH 值7.0~7.5)下的交联。目前羧基反应交联剂的最大缺陷是破坏了蛋白质本身构象,因此在维持蛋白质构象的情况下完成交联是未来羧基反应交联剂的优化方向。
(3)胍基反应的交联剂。精氨酸作为蛋白质中较为丰富的氨基酸之一,大量存在于蛋白质相互作用区域,特异靶向精氨酸胍基的交联剂主要有以乙二醛为反应基团的PDG与BDG,这类交联剂的不足是生成较多不稳定产物,质谱信号分散,检测较为困难[29]。
(4)巯基反应的交联剂。由于马来酰亚胺具有与巯基极高的反应活性,可基于马来酰亚胺设计巯基特异性反应的交联剂,反应时蛋白质中的游离巯基与交联剂的双键发生烷化反应生成稳定的硫醚键,但这类交联剂易发生水解,且对pH值较为敏感,pH值越大越易发生水解[30],可利用它的pH值敏感性,将交联应用到药物与蛋白载体的连接,起到药物在体内定量/定位释放。另外,基于马来酰亚胺和酰肼反应基团的交联剂可适用于连接巯基和糖类,并形成共价交联。
(5)化学切割交联剂。化学切割交联剂是在交联剂连接臂上设计特殊的化学键,化学键可在一定条件下发生断裂,如DSP、DTSSP交联剂连接臂上的二硫键在还原剂作用下断开,分别采集还原前和还原后样品的质谱数据,通过比对还原前后的谱图确定交联肽段。缺点在于还原后得到的两条切割肽段可能无法全部鉴定到,导致交联信息不准确,而且要求体系中不能含与二硫键反应的基团,如自由的巯基等。
(6)质谱切割交联剂。相比于化学切割交联剂,质谱切割交联剂则是在连接臂上设计低能量化学键,如与苯基相连的肽键、固定电荷的硫离子、亚砜和尿素连接的丁酸酯等,这些低能量化学键在二级质谱碎裂时会优先断开实现交联肽对的分离[28]。商品化的DSSO是目前应用较为广泛的一类质谱切割交联剂,它与DSS和BS3具有类似的反应活性,结构简单,两侧的NHS酯基团可与氨基反应,特殊设计在于中间的连接臂插入了亚砜基团,使得两侧对称的C-S键在质谱中优先发生断裂。在二级质谱谱图(MS2)中,DSSO的质谱切割产生特征性谱峰,可根据谱峰识别出交联肽对,再利用三级质谱谱图(MS3)结合搜索引擎进行肽测序得到交联肽段序列。另一种使用较多的质谱切割交联剂是protein interaction reporter(PIR),最早于2005年开发设计,其关键点在于在交联剂中引入不稳定的键,通过质谱的CID、ECD、IRMPD或其它解离机制使交联剂的两个可切割位点原位解离,生成两条肽段与一个报告基团,且切割生成肽段产生的残留基团是常见的修饰,便于检索。根据报告离子与交联肽段的比例关系,识别交联肽段提高鉴定准确性。Hage等[31]引入质谱可切割的“零长度”交联剂:1,1′-羰基二咪唑(CDI),连接蛋白质中的伯胺和羟基基团,其中间臂长度为短羰基单元(~2.6 Å),可以在生理条件(pH 值7.2~8.0)下进行交联反应。质谱切割交联剂简化了蛋白质结构和复杂相互作用的质谱分析,但也存在三级质谱信号强度下降、谱图难以解析的问题。
(7)功能交联剂。可富集的交联剂、同位素标记的交联剂、荧光标记的交联剂、化学选择性连接交联剂以及光反应交联剂等具有特殊功能的交联剂是基于上述交联剂衍生而来的。例如,在交联剂中引入如生物素的可富集基团,使交联肽段检测相对量提高,增加实验结果真实性,甚至在可富集基团设计化学切割位点,实现交联肽段富集后将富集标签去除,减少标签对质谱分析的影响。光反应交联剂主要是利用芳香叠氮、双丫丙啶和其它光反应基团而设计的异型双功能交联剂,可通过两步活化使受体-配体相互作用复合体相关的蛋白、核酸和其它分子发生连接。目前,具有各种特殊功能的交联剂层出不穷,极大地拓展了XL-MS的应用前景,除了用于蛋白质相互作用研究,交联作为一种生物偶联的方式也拓展到了核酸、药物和固相载体表面的修饰。
表1列出了目前常用的部分交联剂。
3.2 质谱数据采集以及谱图解析
由于交联产物两条肽段的结合使搜索空间呈指数级扩大、统计复杂性以及假阳性增加,交联肽段的解析鉴定是XL-MS面临的最大挑战。早期质谱解析是人工对一级质谱谱图(MS1)进行解析:在MS1中找到与理论交联肽段质量相匹配的谱峰,确定其存在,再在MS2的基础上寻找交联位点信息。依据该原理开发的数据解析鉴定软件主要有MS2Links[45]、Links[46]、ProteinXXX[47]、NIH-XL[48]、VIRTUALMSLAB[49]、CLPM[50]、GPMAW[51]、CrossSearch[51]等。但人工解析对MS1的鉴定适用范围小,尤其是当样品复杂时,检索难度急剧上升,很难排除背景噪音、非特异酶切和漏切等因素的影响。
随着质谱技术以及解析软件的快速发展,MS2与MS3的解析鉴定优势逐渐呈示出来,实现了母离子和碎片离子的质荷比信息的同时利用。基于不同化学交联剂(特殊标记、可裂解/不可裂解)引起的交联肽段不同的谱图特点,研究人员针对化学交联剂的特性开发了与之相匹配的数据库搜索引擎,使得交联肽段的谱图信息更加完整,鉴定结果更加可靠。早期出现的针对特殊标记交联剂的搜索引擎:Pro-crossLink[52]、CTUX[53]、Xlink-Identifier[54]、XLink[55]、X!Link[56]、FINDX[57]等可用于简单样品的分析。针对不可裂解的交联剂解析的搜索引擎:xQuest[58]、pLink[24]、pLink 2[59]、Kojak[60]、XiSearch[61]、Protein Prospector[62]和StavroX[63]等均可对大型数据库进行检索。xQuest利用轻重同位素标记的特殊交联剂,第一次尝试了大型数据库搜索。pLink通过对交联肽段碎片离子的特点分析,有效利用内部离子、交联剂断裂离子等的特殊性,提高了交联谱图鉴定率。与需要同位素标记样品的xQuest 相比,pLink可解析无同位素标记和使用特殊交联剂交联的肽段,从而能得到更广泛的应用。pLink 2是一种在蛋白质组规模上准确性和灵敏度更高的搜索引擎,采用两个阶段的开放式搜索策略,将交联肽段看作是一条肽段带有一个未知的修饰,首先通过严格的片段索引,根据得分情况确定一条肽,再利用这条肽段的部分碎片离子进行常规数据搜索。Kojak算法集成了传统数据库搜索算法采用的谱图处理和评分方法,并且可以使用许多不同的带有或没有同位素标记的化学交联剂,可用于分析新的和已有的数据集。XiSearch是针对交联剂BS3的无标记和同位素标记设计的搜索引擎,可使用Skyline软件进行自动化定量,已应用于多种纯化的蛋白质复合物结构分析。此外,Protein Prospector和StavroX均可对复杂的交联数据集进行检索,Protein Prospector软件可用于分析不可裂解交联剂辛二酸二琥珀酰亚胺酯(DSS)产生的交联肽段;StavroX可以在网页上直接进行运算,使用非概率参数来确定交联候选者的得分,可在短时间内解析交联数据。

表1 常用的部分交联剂
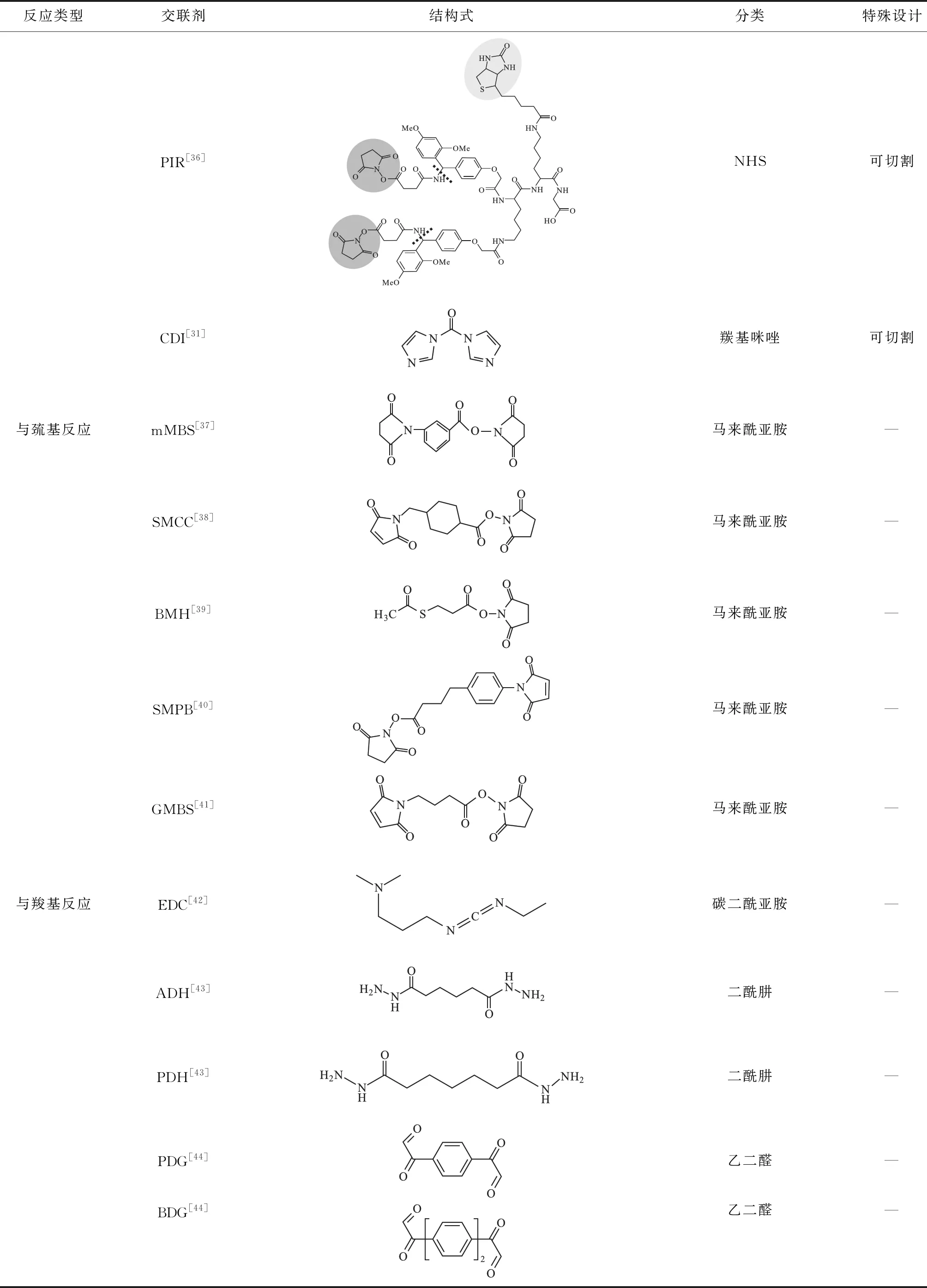
续表1
针对可裂解交联剂的解析方法得益于在MS/MS谱图中可观察到的特定交联报告离子,利用这些报告离子将搜索空间缩小为少数交联肽的组合,目前可裂解交联剂DSBU与DSSO的常用搜索引擎分别为MeroX[64]和XlinkX。MeroX应用于MS可裂解交联剂,扩展了RISEUP和全蛋白质组模式的两种新型算法,可以实现从单个蛋白到全蛋白质组的XL-MS数据分析,它允许在MS和MS/MS两个级别进行质量重新校准,减少了质量匹配和总体计算时间的要求,同时增加了交联肽段鉴定的数量与可靠性。XlinkX 1.0算法首先通过识别所有特征离子峰并找到两个交联肽中每个肽的质量,然后进行基于片段的标准搜索以确定其序列,由于严重依赖于高质量的CID-MS2谱图,研究人员不得不使用复杂的高能碎裂方法进行优化,最大程度识别到特征离子峰。XlinkX v2.0[27]大幅提高了解析蛋白质交联的深度和准确性,它支持MS2-MS3组合的碎裂方法,当MS2裂解交联共价键时,MS3同时将每个交联肽进行裂解,这样就可以在两个水平上提供独特的序列特异性碎片离子,利用这种多样化碎裂优势,XlinkX v2.0可以实现对交联肽段的更全面准确的解析。但这种方法的局限性在于仍需要高强度的特征离子,导致相似键强度的交联剂很难使用该方法区分,影响使用范围。此外,与可碎裂交联剂PIR相匹配的X-links软件[65]和Blinks软件[66]也是常用的检索软件,但它们的缺点主要是MS3时间较长且信号较低,较难捕获解析。
MaXLinker[67]是一种以“MS3为中心”的新型搜索算法,较MS2 有更高的鉴定置信度。虽然XlinkX v2.0、X-Links与Blinks也可进行MS3碎裂分析,但它们基于MS3的交联肽鉴定率与置信度均非常低,MaXLinker基于MS3的算法不仅可以大幅降低错误率,而且提高了交联肽的鉴定率,具有出色的灵敏度和特异性。MetaMorpheusXL[68]是MetaMorpheus软件中一个新的搜索模块,可用于鉴定解析MS可裂解和不可裂解的交联肽。它无需识别交联肽的特征离子,可通过离子索引算法识别MS可裂解的交联肽,从而进行有效的大型数据库搜索。
交联鉴定结果可以通过可视化展示及网络图绘制获得直观分析[28]。最直接的可视化方法是在蛋白质三维结构上直接映射鉴定到的交联位点,但随着鉴定数据的增加,可视化元素和初始输入数据之间没有直接关联,难以检测数据的有效性,为此研究人员开发了许多网络实时可视化的分析软件。Xwalk[69]是一种在已有的晶体结构的交联位点进行模拟计算的软件,通过理论分析交联剂分子性质获得一个较为合理的“溶剂可及表面距离”,计算并显示蛋白质表面化学交联氨基酸之间的曲面距离。XLink-DB[70]是第一个允许编译和分析大规模交联数据的数据库,不仅可以存储、共享和处理交联数据,将交联数据自动映射到PDB结构,使数据可视化并预测蛋白质相互作用,也可将定量蛋白质组数据添加到现有的蛋白质相互作用数据库并进行比较,生成蛋白质相互作用网络,还可以进行物种水平的相互作用分析。Xlink Analyzer[71]是一种在三维结构中分析和使交联数据可视化的工具,通过自动分析交联数据,根据置信度得分与交联类型对数据进行过滤,计算并排除不符合空间约束的交联,绘制蛋白质表面的化学修饰,并且可以将交联映射到同聚寡聚体以及潜在的相互作用位点,与常用的结构显示与建模软件UCSF Chimera交互式综合分析拟合到EM图,获得蛋白质复合物结构的低分辨率模型。xVis[72]能够以圆形、条形图或网络图的形式显示交联位点及距离,可直观描绘出蛋白质区域的空间邻近性以及氨基酸的进化保守性,并根据交联密度排列和分组聚类算法,确定复合物的近端与远端亚基InterPro,利用现有的原位库将轻量级技术和高级数据模型进行整合,最大程度地减少资源浪费[73]。还可根据鉴定分数或错误率筛选交联数据,并显示每条交联肽段相应的碎片离子谱图。xiNET[74]则将视图显示更加直接明确,将交联质谱得到的结果显示为节点图,代表蛋白质之间的相互关系,绘制形成蛋白质相互作用网络。ProXL[75]是一个网页版的应用软件,数据易于共享和导入,支持稳定同位素标记的交联肽段鉴定,可进行精确的质量控制及新颖的可视化分析。Cross-ID[76]不需要文件格式转换,可以直接链接到Proteome Discoverer的XlinkX的输出文件,还可以与其它平台联用实现GO富集分析、翻译后修饰(PTM)可视化、域和二级结构映射、数据集比较等功能分析,通过DisVis在线平台(http://milou.science.uu.nl/cgi/ services/ DISVIS /disvis/)对具有已知结构的蛋白质和复合物或蛋白质复合物进行检测到的交联验证。
4 展望
XL-MS的兴起是传统化学交联方法与现代蛋白质质谱技术相结合发展的结果,对蛋白质复合物结构解析以及蛋白质相互作用研究具有重要作用,“结构决定性质”,了解蛋白质的性质与所承担的功能对生命科学的发展至关重要。但是目前XL-MS的应用仍受到交联剂反应位点具有特异性、交联肽段丰度低、交联肽段谱图复杂、解析困难等诸多因素的限制。新交联剂的设计合成、交联肽段富集分离方法的建立以及交联肽段谱图解析鉴定算法的开发是XL-MS发展的方向。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
辽宁化工(2022年5期)2022-05-28
精细石油化工(2022年3期)2022-05-27
现代仪器与医疗(2022年1期)2022-04-19
食品安全导刊(2021年20期)2021-08-30
现代仪器与医疗(2021年2期)2021-07-21
粘接(2021年2期)2021-06-10
科学技术与工程(2020年3期)2020-04-08
中成药(2019年12期)2020-01-04
分析化学(2018年12期)2018-01-22
