
沉积物磷形态分离分级及其环境地球化学特征
2020-11-17 02:01陈益人
杭州化工 2020年3期
王 毅,崔 健,陈益人
(三川德青科技有限公司,湖北 武汉 430075)
磷被认为是水体富营养化的关键限制因子[1-3],人类的生产、生活活动产生的磷通过地表径流、大气沉降及水气交换作用进入水体后对水体产生严重的污染[4-5],控制磷已成为欧美国家湖泊富营养化的主要管理策略[6]8923,这些策略取得了一定成绩并使一些湖泊的水质得到改善,但也有一些并不成功的案例[7]1014,人们发现这与内源磷的释放有关[8]。 水体磷按照其来源分为外源磷和内源磷,外源磷主要是指高强度的人类活动产生并进入水体的磷,这部分磷经吸附、络合、絮凝和沉降等一系列物理化学过程,最终进入沉积物即形成所谓的内源磷。 当水体向沉积物输入磷的速率大于沉积物向水体释放磷的速率时,即形成内源磷污染的积累,也称沉积物对磷的吸收或负释放;而当沉积物向水体释放磷的速率大于水体向沉积物输入磷的速率时,即形成内源磷的释放[9-12]。 在外源污染被有效阻断后,沉积物通过再悬浮释放等过程仍可对水体形成磷的二次污染[13],从而影响水体水质的恢复并延缓水体富营养化的治理效果[6]8928[7]1014。
因此,对于水体的治理既要关注外源磷污染并对其加以控制,也要重视内源磷污染的释放,要对沉积物中的磷开展深入研究。
1 沉积物磷进行形态分离分级的意义
沉积物主要与水、悬浮颗粒物及生物体之间发生磷的迁移转化,水体中的磷可被悬浮颗粒物中的铁铝氢氧化物和有机质等通过吸附、络合、絮凝并沉降至沉积物,也可通过沉积物- 水界面的交换作用直接被沉积物中的铁锰氧化物、铁铝氢氧化物、黏土矿物、磷灰石和有机质等吸附、螯合及固定,还可直接被生物体吸收并通过生物体的遗体残骸最终沉降至沉积物[14],从而形成沉积物中不同形态的磷,各形态的磷将产生不同的生物有效性及不同的生态学意义[15-16]。
为揭示这些不同的生物有效性及生态学意义,并探索不同磷形态之间的迁移与转化过程[17]403[18],确定沉积物中磷的不同赋存形态显得尤为关键,因此必须开展磷形态的分离分级研究。 在磷形态的分离分级研究中,各形态的磷含量及其分布特征均反映着大量的生物地球化学信息,这对我们认识沉积物磷的生物有效性、生物可利用性以及评价水体的营养状况或污染现状,了解沉积物- 水界面磷的交换作用机制及沉积物内源磷的积累与释放机制,探索成岩过程及评估成岩速率,预防与治理水体磷污染及江河湖库富营养化问题等具有十分重要的指导意义。
2 沉积物磷形态的分离分级方法
2.1 C-J 法
沉积物磷形态的分离分级起源于土壤学中的化学分析方法[19]。1957 年Chang 和Jackson 首次将土壤中的磷分离为不稳定或松散结合态磷(NH4Cl-TP,以1.0 mol/L 量浓度NH4Cl 提取)、铝结合态磷[Al-P,以0.5 mol/L 量浓度NH4F(pH=8.2)提取]、铁结合态磷(Fe-P,用0.1 mol/L 量浓度NaOH 提取)、钙结合态磷(Ca-P,以0.5 mol/L 量浓度HCl 提取)、可还原性水溶态磷[RSP,以0.22/1.0/1.0 mol/L 量浓度柠檬酸钠-连二亚硫酸钠- 碳酸氢钠(CBD)提取]、闭蓄态磷(Oc-P,以0.1 mol/L 量浓度NaOH 提取)和有机磷(Or-P)形态,简称C-J 法[20]。
2.2 W 法
20 世纪六七十年代Williams 等人将C-J 法改进,以0.22/1.0/1.0 mol/L 量浓度CBD 提取沉积物中的非磷灰岩磷(NAP),之后再用0.1 mol/L 量浓度NaOH 提取铁铝结合磷(Fe/Al-P),用0.5 mol/L 量浓度HCl 提取磷灰岩磷(AP),并将沉积物磷主要分为AP、NAP 及Or-P,简称W 法[21-23]。 W 法在提取Fe/Al-P 前未引入F-不会产生CaF2,避免了C-J 法提取的Fe-P 重吸附于沉积物CaF2从而以磷酸铁沉淀的缺陷。
2.3 H-L 法
1980 年Hieltjes 和Lijklema 指出W 法 中部分NaOH 提取的磷可被沉积物中钙盐重吸附的缺点,并提出以0.1 mol/L 量浓度NH4Cl 为提取剂进行第一步提取,在提取不稳定或松散结合态磷的同时去除掉碳酸盐,由此将沉积物磷分为NH4Cl-TP、Fe/Al-P(NaOH-RP,以0.1 mol/L 量浓度NaOH 提取)、Ca-P(HCl-RP,以0.5 mol/L 量浓度HCl 提取)和残余磷[总磷(TP)与各反应性磷总和的差值,该部分磷很难提取,主要以Or-P 形式存在],简称H-L 法[24]。 H-L 法着重于沉积物磷的化学性质分析,有助于认识磷在沉积物-水界面的交换过程及氧化还原电位、离子强度和pH 等环境因子对其交换过程的影响。
2.4 P 法
1984 年Psenner 等人在分析奥地利Piburg Sea沉积物磷形态时提出P 法,该法以H2O、碳酸氢钠-连二亚硫酸钠(BD,0.11 mol/L,40 ℃)、NaOH(0.1 mol/L)、HCl(0.5 mol/L)和NaOH(0.1 mol/L,85 ℃)依次提取水溶 性磷(WSP)、可还原 态 磷(BD-P)、Fe/Al-P、Ca-P 和残渣态磷(Res-P)[25]。Petterson 等人通过对比P 法和H-L 法发现,前者用BD 试剂提取BD-P时存在干扰现象,而后者在用氯化铵2 次提取NH4Cl-TP后会导致HCl 提取的Ca-P 不准确[26]。
2.5 H 法
1995 年Hupfer 等人对P 法进行修改,将第一步的H2O 替换为NH4Cl,依次提取弱吸附态磷(NH4Cl-TP)、BD-P、Fe/Al-P(即图1 中的NaOH-rP)、Ca-P(即图1 中的HCl-P)、有机质结合态磷(NaOH-nrP)和Res-P,即为H 法[27]。 H 法沉积物磷形态分离分级提取流程见图1[28]3386。其中,NaOH-TP 表示NaOH 可提取态磷,为Fe/Al-P、NaOH-nrP 二者之和。 H 法是国际上较为常用的磷形态分离分级提取方法。
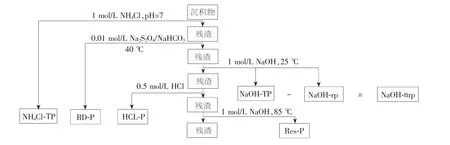
图1 H 法沉积物磷形态分离分级提取流程
2.6 G 法
以往方法中所用的强酸或强碱(如HCl 或NaOH)容易导致Or-P 水解,造成实测值偏高(如Ca-P),因此提取Fe-P 和Ca-P 最好使用螯合剂或络合剂[29]。Golterman 通过采用氨三乙酸(NTA)分别以其钙盐(Ca-NTA)和其钠盐(Na-NTA)提取Fe-P 和Ca-P,有效避免了Or-P 的水解及碱提取引起的重吸附现象,但由于Ca-NTA 在量浓度超过0.02 mol/L 时难溶,因而一次提取的Ca-P 偏低,因此必须重复多次提取,会增加误差[30]。 此外,有些以NTA 提取的尝试并未成功,这可能与NTA 的干扰有关[31-32]。 之后,Golterman等在此基础上发展出了G 法,即用Ca-NTA(0.02 mol/L)和连二亚硫酸钠(0.045 mol/L)的混合液(pH=8)提取Fe-P,而以乙二胺四乙酸二钠(Na2-EDTA)(0.05 mol/L,pH=8)提取Ca-P[33-34]。
2.7 EDTA 法
1996 年Golterman 再次提出用乙二胺四乙酸(EDTA)代替NTA,从而发展形成了EDTA 法,即提取Fe-P 用乙二胺四乙酸二钠钙(Ca-EDTA)(0.05 mol/L,溶剂为1%连二亚硫酸钠,pH=7~8)代替Ca-NTA,并先后分离出Fe-P、Ca-P[以Na2-EDTA(0.1 mol/L,pH=4.5)提取]、酸可提取Or-P(以0.5 mol/L 量浓度的H2SO4提取)和碱可提取Or-P[以2 mol/L 量浓度的NaOH(90 ℃)提取],后者又可分为富里酸磷和胡敏酸磷,在用某些特定的提取剂时还可进一步得到核酸磷、磷酸糖类和肌醇六酸磷等[35]。
2.8 SEDEX 法
1992 年Ruttenberg 在提取海洋沉积物中各形态的磷时提出了一种新的连续提取技术法(SEDEX法),SEDEX 法首次区分了自生钙结合磷与原生碎屑磷(DAP),并以0.1 mol/L 量浓度MgCl2(pH=8)提取交换态磷(Ex-P)、0.3/1.0/0.144 mol/L 的CBD 提取碳酸氟磷灰岩磷(CFAP)、1.0 mol/L 的NaAc/NaHCO3(pH=4)提取Ca-P 和氟磷灰岩磷(FAP)、1.0 mol/L 的HCl 进一步提取出FAP,最后经550 ℃灰化后用1.0 mol/L 的HCl 提取Or-P[36]1461。 SEDEX 法使得沉积物中磷的分离分级适合环境地球化学研究的实际要求,并在每一步提取前均用MgCl2和H2O 进行洗涤以最大限度降低重吸附现象,但该法未考虑提取剂的提取效率不够高。1996 年Baldwin 通过重复每一步提取步骤直至使提取液中磷浓度低于某一阈值对SEDEX法进行改进,使NaHCO3可提取的磷形态在提取4 次后提取效率由60%升至95%[37]。 Kassila 等通过对P 法、H-L 法、EDTA 法和SEDEX 法进行比较,认为EDTA 法优于其他3 种方法,而P 法和SEDEX法对Or-P 会存在低估[38]。
2.9 7 步提取法
1998 年李悦等人将土壤中磷的分离分级提取方法融合至SEDEX 法中,形成所谓的7 步提取法,该法将沉积物磷分成Ex-P、Al-P、Fe-P、Oc-P、自生钙磷(ACa-P)、碎屑钙磷(De-P)和Or-P 等[39]。2004 年朱广伟等对7 步提取法进行改进和简化,如图2 所示,通过采用新鲜沉积物代替风干沉积物样,去掉原方法中每一步用MgCl2溶液与H2O 对残渣进行洗涤的步骤,将Or-P 提取前的灼烧时间从2 h 增加到5 h,并用孔雀绿分光光度法代替钼锑抗测定Ex-P 和Al-P,由此更好地避免了风干等过程造成的沉积物磷的形态变化,并增加了Or-P 提取的稳定性及Ex-P 与Al-P 测定的准确性[40]。 其中,LOI 表示有机质。
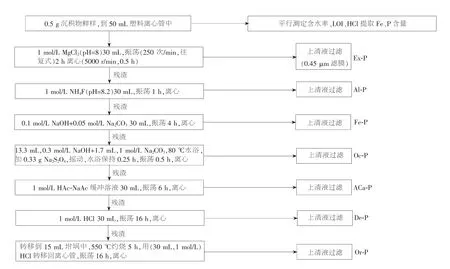
图2 改进的7 步提取法
2.10 SMT 法
沉积物磷形态的分离分级得到快速发展,但并未形成标准的磷形态分离分级方法,针对这种状况,1996 年欧洲标准测试测量组织发起了1 个联合项目,该项目通过对W 法、H-L 法、EDTA 法及SEDEX法等4 种被广泛接受的方法进行对比,在改进的W法基础上形成了淡水沉积物磷形态连续提取的为欧洲标准测试委员会协议(2001年)推荐的SMT 法,SMT 法也是目前较为常用的一种分离分级方法,该法将沉积物磷形态分为Fe/Al-P(即图3 中的NaOH-P)、Ca-P(即图3 中的HCl-P)、无机磷(IP)、Or-P(即图3中的OP)和TP[41]。 Ruban 在对比SMT 法和EDTA 法后认为SMT 法优于EDTA 法[17]403。 SMT 法分析步骤见图3[42]。
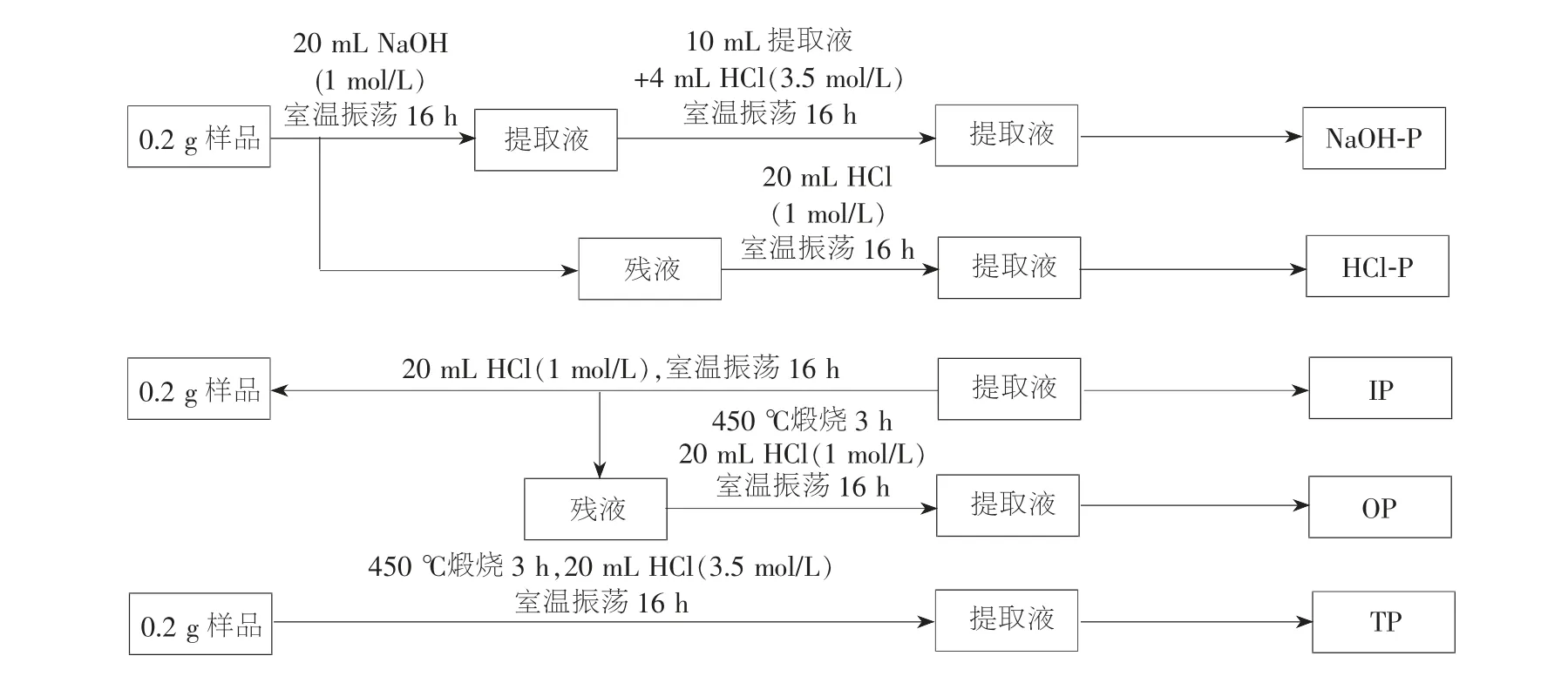
图3 SMT 法分析步骤
2.11 其他方法
2009 年Kovar 分 别 以NH4Cl、NH4F、NaOH、CBD溶液、稀H2SO4溶液和浓H2SO4与HClO4的混合溶液依次提取出NH4Cl-TP、Al-P、Fe-P、RSP、Ca-P 和Res-P[43]。在以往已有磷形态分离分级的基础上,近年来仍有不少学者对其进行改进,如2005 年朱广伟改进的SEDEX 法将沉积物磷分为Ex-P、Al-P、Oc-P、ACa-P 和Or-P 等[44];2018 年高春梅等将沉积物磷分为TP、IP、DAP、Ca-P、Or-P、NH4Cl-RP 及Fe-P[45]。 另外,对于Or-P的进一步分离分级的研究相对较少[46-48],Ivannoff 等将土壤中Or-P 分为活性有机磷(NaHCO3-Po)、中活性有机磷(包括酸提取态有机磷HCl-Po 和富里酸结合态有机磷Fulvic-Po)和非活性有机磷(包括腐殖酸结合态有机磷Humic-Po 和残渣态有机磷Residual-Po)的方法被广泛应用于沉积物中[49]。
3 不同形态磷的环境地球化学特征
磷在沉积物中主要以IP 和Or-P 形式存在,IP是磷参与生物地球化学循环的主要形式[50]115,在SMT 法中,IP 又主要包括Fe/Al-P 和Ca-P[51];Or-P主要指沉积物中动植物的遗体残骸和腐殖质类有机物中包含的磷形态,在微生物或酶的作用下可转化成生物活性磷[52-53]或IP[50]115,从而参与生物地球化学循环,IP 和Or-P 构成TP 的2 个组成部分。 实际上,不同的分离分级方法将得到沉积物中不同的磷形态,以下仅对一些较为常用的方法得到的磷形态的环境地球化学特征进行总结。
3.1 交换态磷
Ex-P 主要指被沉积物中的活性铁锰氧化物、氢氧化物或黏土矿物等颗粒表面吸附或者共沉淀的磷形态[36]1474,该形态磷虽然浓度很低,但却是最易于生物利用的磷形态[54]404。
3.2 弱吸附态磷
NH4Cl-TP 主要系指被沉积物矿物颗粒表面吸附的磷酸盐,也包括孔隙水中溶解性磷[55],是可溶性活性磷酸盐进入沉积物的最初形态,该形态磷是沉积物所有磷形态中含量最低的组分[28]3386,但在沉积物受到扰动或发生其他环境条件变化时,该形态的磷最容易重新进入水体。
3.3 可还原态磷
BD-P 主要是指与铁氢氧化物及锰化合物结合的可还原性磷酸盐[56],该形态磷主要通过化学键的断裂与合成实现转化,受氧化还原电位等环境因子影响较大,具有潜在活性,可为藻类所利用,该形态作为评价沉积物内源磷复核的重要指示指标[57]。
3.4 铁铝氧化态磷
Fe/Al-P 主要为Fe、Al 金属氧化物结合的磷[58],一般公认是沉积物中较为稳定形态磷的组成部分,属于潜在活性的磷,该形态磷受pH 影响较大,在pH 较高时由于OH-与磷酸根发生置换能形成更稳定的Al(OH)3或Fe(OH)3,会导致磷酸根的释放,从而易于被生物利用[59]。 Fe/Al-P 主要包括Fe-P 和Al-P,其中Fe-P 是磷与铁氧化物或氢氧化物发生共沉淀形成的磷酸盐[14]3,该形态磷可以被生物(如藻类等)所利用,在一定条件下(如温度或溶解氧发生变化[60])容易释放至上覆水中[54]405;Al-P 是磷与铝氧化物或氢氧化物结合的磷形态,该形态磷易与溶解的磷酸盐复合物交换或与OH-发生反应而溶解,后者将形成既可以在好氧条件又可以在厌氧条件下稳定存在的Al(OH)3。
3.5 有机质结合态磷
NaOH-nrP 主要是用NaOH 提取的Or-P 及腐殖酸磷[61],该形态磷主要通过NaOH 提取到的TP 减去Fe/Al-P 得到,该形态磷通过细菌矿化分解后可成为内源磷的重要来源[62],有机质因其附着作用成为该形态磷的载体,其对沉积物磷的固定及该形态磷的含量产生很大的影响[63]。
3.6 钙结合态磷
Ca-P(HCl-P)主要为磷灰石磷,其包括与碳酸盐结合的磷和微量可水解的Or-P,是沉积物磷中相对稳定的组分,也是磷在沉积物早期成岩的最终产物之一,该形态有利于磷在沉积物中的长期埋藏,但在酸性环境中该部分磷可以释放出来。Ca-P 按照其来源可以分为ACa-P 和De-P,前者主要指包含于沉积物中的原生矿物颗粒中的钙磷,该部分钙磷主要来源为各种难溶性的磷酸钙矿物(如过磷酸钙等),该形态钙磷稳定性比较高,较难被生物利用[64]57;后者主要指沉积物中通过生物作用沉积或固结的颗粒磷(如羟基磷灰石等),该部分钙磷主要反映了动植物遗体残骸引入沉积物的磷[54]405,同样难以被生物利用[64]57。
3.7 闭蓄态磷
Oc-P 主要为铁的氧化物或铁铝矿物包裹的还原性磷酸铁或磷酸铝,还包括一部分硅酸盐晶格中的磷,该形态磷可能来源于风化作用,或早期成岩过程中形成的含磷自生矿物,该部分磷属于稳定态[65],即使在强还原条件下也很难释放出来。
3.8 残渣态磷
Res-P 也称为惰性磷,主要为禁锢在矿物和其氧化物晶格中的磷[66],该形态磷在环境条件发生改变时也基本不会释放出来,是沉积物所有磷形态中最为稳定的部分[67]。
4 展望
尽管前人对于磷的分离分级已经进行了许多较为深入的研究,但至今仍未形成一种国际或国内通用的沉积物磷的分离分级方法,许多研究数据缺乏可比性,很多方法仍是基于操作上的便利而非对于具体化学存在形态或结构上的研究,并且很多方法均存在一定的缺陷性。 对于Or-P,其形态更是复杂多样和高度可变,许多新的方法和高端技术如X 射线衍射技术、核磁共振等逐渐应用到磷形态的研究中。
展望未来,加快对磷形态分离分级技术的研究,改进前人方法的不足,并借助高端技术从分子和化学结构角度对沉积物磷形态进行探索和提高分析结果的准确度仍是磷形态分离分级研究的首要任务;另外,运用单一的分离分级方法对沉积物磷形态进行定量分析时需谨慎,最好通过多种被广泛接受的方法综合比较再作结论;此外,仅从环境领域研究磷形态的分离分级存在较大的局限性,加大如流体力学、物理学、化学、矿物学和水力学等学科领域的交叉与结合研究,势必能解决以往所遗留的一些科学难题。 经过不断探索和不断更正,建立磷形态分离分级方法的标准体系是我们的重要目标。 通过一系列关于磷形态的研究,总结经验并应用于水体磷污染的治理与管控是我们的最终目标。目前,对于Fe-P 的研究已提出通过增氧曝气和纳米气泡矿物改性颗粒等技术控制沉积物-水界面溶解氧从而控制内源磷的释放,这些技术和经验的发展增加了我们对于磷形态分离分级研究的信心,未来势必可期。
猜你喜欢
江苏农业科学(2022年15期)2022-08-11
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
世界最新医学信息文摘(2021年12期)2021-06-09
兽医导刊(2020年13期)2020-12-31
昆明冶金高等专科学校学报(2020年1期)2020-07-06
农业环境科学学报(2019年12期)2019-12-23
农药科学与管理(2019年7期)2019-11-29
江苏农业学报(2019年2期)2019-09-10
蔬菜(2019年5期)2019-01-04
