
SAPO-5分子筛的晶化过程研究
2020-11-13 09:37罗东霞王全义刘仲毅刘中民
高等学校化学学报 2020年11期
罗东霞,李 冰,王全义,田 鹏,刘仲毅,刘中民
(1.郑州大学化学学院,绿色催化研究中心,郑州450001;2.中国科学院大连化学物理研究所甲醇制烯烃国家工程实验室,洁净能源国家实验室,大连116023)
SAPO分子筛作为分子筛家族的重要成员已经在催化、吸附和离子交换等领域展现了优异的性能[1~3].对SAPO分子筛的晶化机理进行研究有助于增强对晶化过程的调控能力.同时,由于硅的进入使得SAPO分子筛生成具有催化性能的酸性位,开展硅进入分子筛骨架及其在晶体中分布的研究有助于深入认识SAPO分子筛的酸性,并实现对酸性的理性调控.
SAPO-5分子筛的骨架结构为AFI型,具有平行于[001]方向的一维十二元环孔道,孔口尺寸为0.73 nm×0.73 nm(图1).SAPO-5分子筛主要采用水热法合成,多种有机胺均可作为模板剂导向SAPO-5 的生成[4~6]. 此外,微波合成法[7~9]、两相合成法[5,10,11]、溶剂热合成法[12,13]及干胶法[14,15]等也被用于SAPO-5分子筛的合成.

Fig.1 Structure(A)and channel system(B)of SAPO⁃5 molecular sieve
晶化机理方面的研究多集中在AlPO4-5分子筛.Thomas等[15]采用原位能量色散X射线衍射(EDXRD)追踪了AlPO4-5和CoAlPO4-5分子筛在晶化过程中的结构演变;研究发现,前者经历从诱导期到AlPO4-5晶体形成的过程,而后者在诱导期后会同时形成AFI和CHA晶相,CHA随着晶化过程的进行会消失.该结果也说明凝胶中钴离子的存在会改变晶化过程.Deng等[16]采用多种固体核磁技术对含氟体系AlPO4-5分子筛的晶化过程进行了研究,发现五配位的Al物种单元F-Alpen-O-P(Alpen表示五配位铝物种)的出现标志着成核和晶体生长的开始;无定形初始凝胶中的F-Alpen-O-Ppar和Alpen-O-Ppar(Ppar表示部分缩合的磷物种)逐渐转化为骨架中的F-Alpen-O-Pful和Altet-O-Pful(Altet表示四配位铝物种;Pful表示完全缩合的磷物种).Huang等[17]利用固体核磁共振波谱结合X射线衍射(XRD)及拉曼光谱方法监测了Al-PO4-5分子筛的水热晶化过程,发现磷源和铝源在模板剂存在下混合后即生成了无定形磷酸铝及磷酸铵,加热后无定形结构快速重组,多数四配位铝先转变为八面体铝,磷酸盐物种被“溶解”,直接从无定形材料晶化得到AlPO4-5.Huang等[18]还研究了干胶转化法合成AlPO4-5分子筛的晶化过程,发现晶化过程中首先形成了具有AFI骨架结构特征的前驱体,并通过这种前驱体的水解和缩合最终生成了AlPO4-5.Li等[19,20]用原位拉曼光谱研究了AlPO4-5分子筛的水热合成机理,发现晶化初期先生成了四元环链状结构,之后链状结构重组生成无定形类孔道结构,再由这些无定形类孔道结构重排得到最终的产物AlPO4-5分子筛.Hu等[21]利用原位固体核磁技术表征了AlPO4-5分子筛的晶化过程,揭示了磷、铝物种初步缩聚后以Al—O—P链形式存在,在模板剂和水的作用下通过重排缩聚形成四/六元环交替的半晶化链状结构,并通过进一步缩合形成具有十二元环孔道的骨架结构.Xiao等[22]对干法合成AlPO4-5的晶化过程进行了研究,发现初始的无定形原料首先转化成含有四/六元环的中间相,然后再逐渐转变成AlPO4-5.
目前,关于SAPO-5晶化过程的详细研究较少.一方面由于硅的存在可能使分子筛的晶化过程与AlPO4-5存在区别;另一方面,对硅在分子筛骨架中的配位环境随晶化时间变化进行研究有助于增加对SAPO-5分子筛酸性质的认识和调控.因此,本文采用XRD、X射线荧光光谱(XRF)、扫描电子显微镜(SEM)、固体核磁共振波谱(MAS NMR)和X射线光电子能谱(XPS)等表征手段对SAPO-5分子筛的水热合成过程进行了研究.
1 实验部分
1.1 试剂与仪器
拟薄水铝石(Al2O3,质量分数为67%)购于山东铝业股份有限公司;硅溶胶(SiO2,质量分数为27.5%)购于沈阳新光脱膜剂厂;磷酸(H3PO4,含量85%)、正硅酸乙酯(TEOS)和三乙胺(C6H15N,TEA)均为分析纯,购于天津市科密欧化学试剂有限公司;去离子水(自制).
荷兰PANalytical公司X′Pert PRO型X射线衍射仪(CuKα辐射源,λ=0.15418 nm,管电压40 kV,管电流40 mA,扫描范围5°~50°);中国科学院科学仪器厂KYKY-AMRAY-1000B型扫描电子显微镜;荷兰Philips公司Magix-601型X射线荧光光谱仪;美国Thermo Fisher Scientific公司ESCALAB 250Xi型X射线光电子能谱仪[以Al/Mg双阳极为激发源,以样品表面Al2O3的Al2p(74.7 eV)为内标来校正样品表面的荷电];瑞士布鲁克公司Bruker AvanceⅢ600(14.1 Tesla)型核磁共振波谱仪.
1.2 实验过程
SAPO-5分子筛的制备:按P2O5/Al2O3/SiO2/TEA/H2O摩尔比为1.0∶1.0∶0.4∶1.2∶60,将磷酸、拟薄水铝石、TEOS与去离子水混合,室温搅拌均匀后,加入模板剂TEA,继续搅拌至均匀.将所得的凝胶转移至2 L内衬为聚四氟乙烯的不锈钢反应釜中,密封,程序升温至晶化温度(200℃)后恒温晶化.升温即开始计时,在不同时间点从釜中在线取样.在取样时间分别为0.92和1.5 h时,对应的温度分别为100和150℃,而取样时间为1.83 h及以后时间的温度均为200℃.取出的样品冷却至室温,然后在没有外加水的情况下直接离心分离得到母液和固体样品.母液于120℃烘干后于550℃下焙烧并测定其固体含量.固体样品用去离子水洗涤至中性后于120℃干燥.
以硅溶胶为硅源的SAPO-5分子筛的制备与上述过程类似,不同的是用硅溶胶代替TEOS.凝胶中P2O5/Al2O3/SiO2/TEA/H2O摩尔比为1.0∶1.0∶0.4∶1.2∶60.将所得凝胶移入100 mL内衬为聚四氟乙烯的不锈钢反应釜中,密封,程序升温至200℃,晶化24 h.
2 结果与讨论
2.1 XRD表征
图2是不同晶化时间固体样品的XRD谱图.可见,在晶化的初始阶段(1.83 h之前),由于晶化时间短且温度较低,固体样品主要是未晶化的无定形一水软铝石胶[23].晶化温度达到200℃后(1.83 h)之后,在7.4°,14.9°,19.7°,21°和22.3°等位置呈现出SAPO-5分子筛的特征衍射峰,表明SAPO-5晶相开始生成.此后,随着晶化时间延长,所得固体产物的衍射峰位置不变,但峰强度增强.

Fig.2 XRD patterns of the as⁃synthesized samples with different crystallization time

Fig.3 Crystallization curve of SAPO⁃5
以SAPO-5的XRD谱图中4个最强峰的峰面积和为基准,计算不同晶化时间样品的相对结晶度(18.83 h样品结晶度计为100%),得到SAPO-5分子筛的晶化曲线(图3).在晶化时间为3.33 h之前,样品相对结晶度随着晶化时间延长快速增加,2.33 h时样品的相对结晶度已经达到67%;晶化时间为3.33 h时相对结晶度为83%;此后随着晶化时间延长,相对结晶度持续增加,至18.83 h达到最大值.
2.2 SEM表征
图4是不同晶化时间样品的SEM照片.在晶化时间为0.92及1.5 h的样品中只能看到无定形物质;而晶化时间为1.83 h的样品中仅含有少量无定形物质,除少量六方形薄片或柱状SAPO-5外,存在大量由薄片构成的球状聚集体(记为物质Am).随着晶化时间的延长,SAPO-5晶体尺寸逐渐均匀,含量增加,薄片构成的球状物含量降低,但直至晶化结束,样品中仍能观察到少量球状聚集体的存在.结合前述的XRD结果可知,物质Am尽管具有相对规整的形貌,但不具有长程有序结构.进一步采用SEM-EDX对物质Am的组成进行了分析,显示其组成主要为磷铝,仅含极少量的氧化硅.

Fig.4 SEM images of the as⁃synthesized samples with different crystallization time
2.3 XRF分析
采用XRF检测了不同晶化时间样品的元素组成.图5示出了固体样品中P,Al和Si 3种元素的相对摩尔分数随晶化时间的变化结果.可见,在2.33 h之前,固体样品中的铝含量呈现快速下降趋势,由0.92 h的66.8%减少为2.33 h的49.3%,表明随着晶化的进行,来自原料中的铝物种不断溶解进入液相;磷含量在1.5 h时出现极小值,由0.92 h的29.4%减少为22.3%,在1.5 h之后快速上升,至2.33 h时增加至45.7%;硅含量的变化则与磷相反,其在1.5 h呈现一个极大值.结合XRD和SEM结果认为,1.5 h后固体样品中磷含量的增加应该与SAPO-5分子筛和物种Am的生成相关,硅含量在1.5 h时的最高值则有可能与TEOS原料的大量水解生成聚合态氧化硅有关,之后氧化硅逐渐溶解进入液相.在0.92 h样品的离心母液中可以明显观察到分层的TEOS存在,显示此时TEOS还未水解完全,而1.5 h的母液则为均一相,这与XRF分析的组成变化一致.随着晶化时间的进一步延长,固体样品中的铝磷呈现微弱的下降趋势,至晶化时间为18.83 h时,样品中铝的摩尔分数为48.8%,磷的摩尔分数为44%.此阶段硅的相对含量呈增加趋势,其变化幅度大于磷铝2种元素,2.33 h时样品中硅的摩尔分数为5.1%,18.83 h时增长了1.41倍(7.2%).
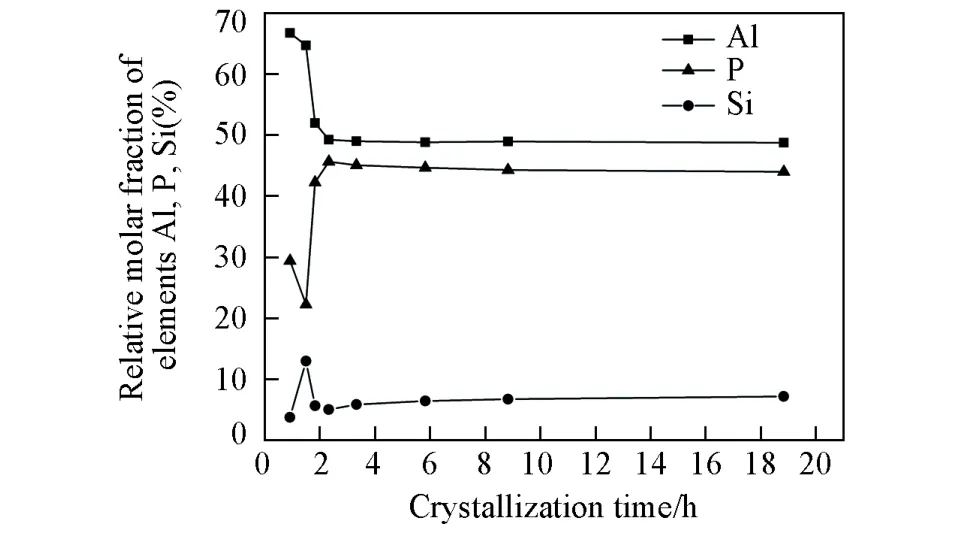
Fig.5 Chemicalcomposition ofas⁃synthesized samples with different crystallization time

Fig.6 Solid content(a)and pH value(b)of the mother liquids with crystallization time
2.4 母液固体含量及pH值
图6谱线a显示了母液中固体含量(质量分数)随晶化时间的变化趋势.可见,在晶化初期,母液固体含量有一个明显的波动过程,即从0.92 h的11.5%增加为1.5 h的13.2%,之后母液固体含量快速降低,至3.33 h时达到9%.此后随着晶化时间延长固体含量逐渐降低,晶化结束时为5.7%.结合XRD及XRF结果表明,晶化前期固体含量极大值的出现应该与原料的溶解有关,而之后的降低则主要是由于分子筛的逐渐生成引起的.从SEM结果可知,合成体系后期固体含量的变化应该是分子筛生成和物质Am溶解相互平衡的结果,但总体上,SAPO-5的生成量要大于物质Am溶解的量.
母液的pH值与合成体系中各物料状态的变化密切相关.由图6谱线b可知,在3.33 h之前,母液的pH值随晶化时间的延长快速增加.之后至晶化结束,母液的pH变化较小,基本维持在7.3左右.结合上述实验结果可以认为,晶化初期至1.83 h期间合成体系pH值的快速增加是由于磷酸与氧化铝反应而被消耗所导致的(促进了氧化铝原料的溶解),之后随着分子筛的逐渐生成(对应于母液固体含量的降低),溶液pH值进一步上升,至5.83 h时pH值达到7.4;随后至晶化结束pH值仅发生微小波动.
2.5 固体MAS NMR表征
不同晶化时间固体样品的31P MAS NMR和27Al MAS NMR谱分别见图7(A)和(B).图7(A)中晶化时间为0.92 h的样品只有一个中心位于δ—16的宽包峰,可归属为无定形磷酸铝相中的磷物种[24].对应的27Al MAS NMR谱在δ8(强)和δ—8(弱)处有2个共振峰,可分别归属为无定形铝源中的五配位铝物种和无定形磷酸铝中六配位铝物种.另外,δ8的共振信号也有可能包含无定形磷酸铝中少量存在的五配位铝物种的贡献[25,26].晶化1.5 h后,图7(A)上除了宽包峰外,在δ-19附近出现了一个尖锐的小肩峰,显示无定形固体中出现了具有较高缩合度且有序度较高的磷物种[25].对应的27Al MAS NMR谱中除六配位铝物种信号强度有一定增加外,变化不大.晶化进行1.83 h后,图7(A)中对应于无定形磷酸铝相的宽包峰消失,δ-19处的峰明显增强,同时在δ-30处出现一个可归属为骨架四配位磷物种的信号[27].对应的27Al MAS NMR谱在δ-10.5处的峰明显增强,δ8处的峰减弱,同时在δ37.7处出现1个新的共振峰.δ8处共振信号的降低说明样品中无定形氧化铝含量降低,而δ37.7的信号则对应于SAPO-5分子筛的生成,可归属为骨架四配位的铝物种[23].另外,δ-10.5的共振信号有可能来自SAPO-5的骨架六配位铝物种[28,29],强度与通常的SAPO-5样品有所不同,说明有可能存在其它情况.进一步延长晶化时间至晶化结束,磷铝核磁共振波谱中相应于骨架四配位的信号峰强度均明显增强,说明它们成为固体样品中的优势磷铝物种,而图7(A)中δ-19和图7(B)中δ-10.5处的信号强度则相应降低.结合SEM表征结果,自1.83 h起存在大量无定形物质Am,其随晶化时间延长而逐渐减少,这与磷铝核磁共振波谱中δ-19和δ-10.5处的信号强度变化趋势一致,推测这2个信号应该与物质Am有关.

Fig.7 31P(A)and27Al(B)MAS NMR spectra of the as⁃synthesized samples with different crystallization time
图8是不同晶化时间固体样品的29Si MAS NMR 波谱.可见,0.92 h样品的谱图在δ-112处存在一个可归属为无定形氧化硅中Si(OSi)4环境的强共振信号[26],同时在δ-94.3附近还有一个较宽的弱肩峰,显示样品中存在具有较低缩合度的硅物种[Si(OSi)2(OH)2].延长晶化时间至1.5 h,δ-112的峰向高波数发生位移,同时肩峰的相对强度也有一定增大.晶化至1.83 h时,硅谱在δ-93.2处开始出现一个比较尖锐的信号.结合XRD、SEM-EDX(无定形物质Am主要由磷铝组成)和磷铝核磁共振波谱结果可知,此时样品中开始出现SAPO-5分子筛,这里将该信号归属为SAPO-5分子筛中的Si(4Al)物种.但是需要指出的是,δ-108处强共振峰的存在显示此时固体样品中的硅多数仍以无定形氧化物的形式存在.晶化时间3.33 h样品的硅谱中分子筛骨架Si(4Al)物种的信号明显增强,同时在δ—98.1,δ—102.3,δ—105.7和δ—109处出现交叠在一起的宽共振信号.考虑到此时固体样品中主要存在SAPO-5晶体和无定形物质Am(磷铝组成)(图4),同对将后面的共振峰依次归属为骨架的Si(3Al),Si(2Al),Si(1Al)和Si(0Al)物种[30],该结果同时也说明3.33 h样品中的硅岛物种的含量较高.XRF表征结果显示,3.33 h样品中硅的相对摩尔分数为6%,说明SAPO-5的骨架中仅能容纳较低含量的Si(4Al)物种.进一步延长晶化时间,δ-112处对应于硅岛物种的信号强度有所降低,Si(4Al)物种的含量增加.

Fig.8 29Si MAS NMR spectra of the as⁃synthesized samples with different crystallization time
2.6 晶化过程分析
根据前面的表征结果,对SAPO-5分子筛晶化过程分析如下:晶化进行0.92 h(对应升温到100℃)时,大量的磷源进入液相,少部分的磷以无定形磷酸铝的形式存在于固相;铝源主要存在于固相,其中少量铝源和磷酸反应生成了无定形磷酸铝;多数硅源以原料形式存在,仅少部分水解后聚合沉积在固相.晶化1.5 h(对应150℃)后,磷源和铝源进一步溶解进入液相,TEOS水解完全并主要以无定形氧化硅形式存在于固相,尽管XRD显示固体样品为无定形,但起始阶段样品中已经开始出现具有一定有序度的高缩合度磷酸铝物种Am.晶化温度升高至200℃(晶化时间为1.83 h)时,无定形物质Am的量明显增加,同时SAPO-5开始生成,此外固相中还包含少量未反应的铝源和无定形氧化硅.进一步延长晶化时间至晶化结束,无定形物质Am逐渐溶解,SAPO-5的结晶度和收率逐渐增加.硅从晶化初期即直接参与SAPO-5骨架的形成,其在SAPO-5晶体中的含量随着晶化时间的延长而增加.另外,硅核磁共振波谱结果显示SAPO-5骨架仅能容纳较低含量的Si(4Al)物种,分子筛中硅摩尔分数约为6%时,已经存在大量的硅岛环境.
进一步采用XPS分析SAPO-5晶体的表面元素组成,结果列于表1.很明显SAPO-5的表面硅含量高于体相,表明硅在SAPO-5晶体中的空间分布是不均匀的,呈现从内向外递增的趋势.这与之前关于SAPO-34和SAPO-35的研究结果[23,31]一致,显示SAPO分子筛晶体表面富硅现象具有一定的普遍性.不同结构的SAPO分子筛的表面富硅程度差异可能与骨架结构和合成条件有关.表1同时给出了使用硅溶胶作为硅源合成的SAPO-5(Si sol)的体相和表面元素组成.可见,尽管晶化时间为18.83 h样品和SAPO-5(Si sol)具有相同的初始凝胶摩尔比,但以TEOS为硅源合成得到的晶化时间为18.83 h样品具有更高的硅含量,说明TEOS比硅溶胶具有更高的反应活性,更易进入分子筛骨架.此外,前者具有相对较低的表面硅富集程度(R值),显示合成使用的硅源会影响分子筛晶体的表面富硅程度.

Table 1 Bulk and surface compositions of SAPO-5
3 结 论
采用多种表征手段研究了SAPO-5分子筛的水热晶化过程,发现SAPO-5的晶体生长过程符合液相转变机理.晶化初期(晶化时间小于等于1.5 h),固相/液相的元素组成、液相的固体含量及液相pH值均发生了剧烈变化,大量的磷源和部分铝源溶解进入液相;固相中则包含未反应的铝源、无定形磷酸铝及无定形氧化硅(TEOS水解得到);晶化时间为1.5 h的样品中开始出现具有较为规整形貌的无定形磷酸铝物质Am.晶化温度升高至200℃时(晶化时间为1.83 h),无定形物质Am的量明显增加,同时SAPO-5开始生成.延长晶化时间至晶化结束,无定形物质Am逐渐溶解,SAPO-5的结晶度和收率逐渐增加.硅从晶化初期即直接参与SAPO-5的骨架形成,其在晶体中含量随晶化时间的延长不断增加.SAPO-5分子筛骨架仅能容纳少量的Si(4Al)环境,在样品中硅摩尔分数约为6%时已经出现大量的硅岛环境.此外,SAPO-5分子筛存在表面富硅现象,硅在晶体中分布不均匀,从核到壳含量递增.
猜你喜欢
辽宁化工(2022年8期)2022-08-27
建材发展导向(2021年13期)2021-07-28
中国材料进展(2020年4期)2020-05-23
硅酸盐通报(2020年1期)2020-02-25
石油炼制与化工(2020年2期)2020-02-20
陶瓷学报(2019年6期)2019-10-27
火工品(2019年3期)2019-09-02
商品混凝土(2019年6期)2019-07-02
西北药学杂志(2018年5期)2018-09-20
中国氯碱(2018年5期)2018-06-01
