
DNA酶:筛选、生物传感及展望
2020-11-13 09:37佟宗轩胡沁沁顾宏周
高等学校化学学报 2020年11期
佟宗轩,胡沁沁,顾宏周
(复旦大学附属中山医院心血管疾病研究所,复旦大学生物医学研究院,上海200032)
DNA酶又称为脱氧核酶(Deoxyribozymes,DNAzymes),通常由一定长度(几十个碱基)和特定序列的单链DNA通过折叠成某些二维和三维空间结构而获得催化功能.第一个DNA酶出现在1994年,Breaker等[1]利用指数富集的配体进化技术(SELEX),在试管内首次获得了可以在特定位点切割核糖核酸(RNA)的脱氧核糖核酸(DNA)序列[图1(A)],这项工作揭开了DNA酶的面纱.此后的系列研究发现,DNA酶可以像RNA酶(Ribozyme)和蛋白酶一样高效催化DNA/RNA连接、DNA磷酸化、DNA腺苷化、DNA脱嘌呤化及狄尔斯-阿尔德反应等生化反应[2~6].从化学的角度来看,DNA只比RNA少了1个2′-OH,其它化学官能团几乎完全相同,因此DNA像RNA一样具备多种催化功能.与RNA酶和蛋白酶相比,DNA酶具备一些天然的优势:因其构成为DNA,故稳定性极佳,且易于化学合成修饰、易于操作和储存且制备成本低. 这些优势推动了其在生物传感[7,8]、基因治疗[9~11]和生物成像[12,13]等领域的发展与应用.尽管已有数十种DNA酶在试管内进化分离出来,但迄今尚无很强的证据显示DNA酶在自然界中也存在.
除了一类可催化RNA连接反应[14]和一类可催化RNA切割反应[15]的DNA酶的工作机理在原子精度上得到了阐释之外,目前绝大多数DNA酶的工作机理尚未知,但DNA酶的催化方式有一定的共性.通常,DNA酶由非保守序列区和保守序列区两部分组成,前者的主要功能是通过碱基互补配对识别并作用于底物,在这个过程中后者与底物之间的距离拉近并可通过进一步折叠形成特定结构以催化底物特定位点上的反应.以切割RNA的第一个DNA酶为例[图1(A)][1]:两端的碱基互补配对使得DNA酶与RNA底物序列间形成很好的识别和杂化,而酶上loop区的保守序列在Mg2+的帮助下折叠成特定的具有催化活性的结构,此结构可与RNA底物上切割位点处的碱基发生二级和三级相互作用,激活RNA碱基上的2′-OH作为亲核试剂攻击邻近磷酸二酯键上的5′-O,从而切断RNA底物链上的磷酸二脂键.通过碱基互补配对识别底物的方式使DNA酶具有很强的可编程性,但同时也带来了多重催化效率低下(与自然界存在的蛋白酶相比)的问题:在单个催化转换(single-turnover)效率中,较强的杂化才能使DNA酶较好地识别底物,从而高效地完成single-turnover反应;但较强的杂化作用使反应完成的底物难以从DNA酶上脱落,成为阻止DNA酶继续捕获未反应的底物催化多重催化转换(multiple-turnover)的障碍.最近,我们[16]发现可以通过简单的升降温来促进DNA酶与底物间的分离/结合,从而使DNA酶的multiple-turnover效率在体外提升至可以与蛋白酶的效率相媲美[图1(B)].尽管该升降温的策略主要是在切割DNA的DNA酶上得以验证,但理论上其应当可适用于提升绝大多数DNA酶的turnover效率.

Fig.1 Deoxyribozymes and the classical selection strategy to isolate them
目前,几乎所有已知的DNA酶都是通过SELEX技术体外筛选分离获得的,经典的DNA酶的筛选策略主要由合成/制备文库、与辅助因子共孵育、分离信号DNA序列及聚合酶链式反应(PCR)扩增再建库等4个步骤的多轮次循环往复构成[图1(C)].以切割RNA的DNA酶的筛选为例进行说明.首先,化学合成和制备含有随机序列区、引物结合区及一个核糖核苷酸(rA)为假定切割位点的初始文库.该文库一般包含约1014~1015个DNA序列,文库5′端修饰生物素标签,可通过生物素-链霉亲和素的相互作用锚定在固定相上.当辅助因子(如金属离子、小分子和生物样本等)与文库共孵育时,如果某些DNA序列可以与辅因子相互作用折叠成活性结构切割rA位点,则切割后其将从固定相中脱落释放出来;而无活性未发生切割的DNA序列则仍保留在固定相上.切割脱落的DNA序列经两步PCR扩增后,重新引入rA切割位点和生物素标签,用于生成次级文库进行下一轮筛选.通常,上述循环过程需重复5~20轮,并可通过缩短孵育时间及降低辅助因子浓度等方式施加筛选进化的压力.具有切割RNA活性的DNA序列逐步被富集,在文库中所占比例也不断升高,经过测序和实验验证进而明确最优的DNA酶的序列信息.
迄今,利用经典的筛选策略已分离出多种DNA酶.随着技术的进步和下游应用领域的需求不断增加,DNA酶的筛选策略也在不断发展和更新.本文将回顾近5~10年内涌现出的筛选新策略及依托其分离出的新型DNA酶,重点阐述响应金属离子、小分子及生物样本(包括细菌、细胞或其提取物等)并切割RNA的DNA酶探针,并展望本领域现存的挑战和发展方向.
1 催化多种生化反应的DNA酶
1.1 产物捕获的筛选策略
早期发现的DNA酶大多作用于核酸底物,包括RNA 切割酶[1,17,18]、DNA 连接酶[19]、RNA连接酶[20,21]、DNA切割酶[22,23]及DNA磷酸化酶[24]等. 选择核酸作为DNA酶作用底物的主要原因在于两者属于同一类材料,有利于核酸筛选文库的构建和核酸信号的扩增放大.近年来,研究人员更多地将目光投向蛋白质/多肽的化学催化反应,通过设计新型筛选策略,分离出催化各类蛋白/多肽生化反应的DNA酶,为进一步拓展DNA酶的应用奠定了基础.
通常,蛋白质/多肽生化反应仅仅是化学基团增减的细小变化(如磷酸化、去磷酸化修饰等).如何判断DNA序列确实催化了这些生化反应,并有效地从文库中分离出具有催化活性的DNA序列,是成功筛选可催化蛋白质/多肽生化反应的DNA酶的关键.为此,Silverman等[25]开发了产物捕获的筛选策略(PCS):在每一轮筛选过程中,用额外的分子(如抗体或修饰的核酸等)与具有所需催化功能的DNA序列发生交联反应或结合,将其捕获,再通过简单有效的凝胶电泳技术将其与无催化活性的DNA序列分离.运用PCS方法,已成功筛选出多种催化蛋白/多肽生化修饰反应的DNA酶.
2013年,Silverman等[26]报道了第一个具有酪氨酸激酶活性的DNA酶.该DNA酶以5′-三磷酸化RNA寡核苷酸(5′-pppRNA)为供体,可将γ-磷酸基团转移到多肽链中酪氨酸的羟基上,使酪氨酸(Tyr)磷酸化(即变为pTyr).在关键的捕获步骤中,Silverman等使用固相抗pTyr抗体来分离每轮具有催化活性的DNA序列[图2(A)].与凝胶电泳等方法相比,固相抗体策略显著缩短了筛选时间,且可实现多样品的同时处理.在后续工作中,Silverman等[27]以不同的天然多肽序列为底物,利用固相抗体捕获策略高效分离出一系列可催化特定多肽序列中酪氨酸磷酸化的DNA酶,这进一步验证了该策略的普适性,并为开发针对生物样本催化酪氨酸磷酸化的DNA酶扫清了障碍.
与磷酸化相对应,Silverman等[28]同时探索了DNA酶催化氨基酸的去磷酸化反应.在筛选过程中,首先构建了包含随机序列和偶联六肽底物(AAASPAA)的核酸文库,随后采取产物捕获策略,即通过5′-硫醇寡核苷酸与脱氢丙氨酸(Dha)发生迈克尔加成反应,增加有催化活性DNA序列的分子量,即可利用凝胶电泳将其与无催化活性的核酸序列进行有效分离[图2(B)],捕获效率可达40%.研究结果表明,在Zn2+或Zn2+/Mn2+的辅助下,分离得到的DNA酶可有效去除磷酸化丝氨酸(pSer)中的磷酸基团,生成脱氢丙氨酸(Dha).与催化氨基酸磷酸化的DNA酶一起,Silverman等的研究[26~28]为DNA酶在磷酸化蛋白质组学方面的潜在应用打下了基础.
蛋白质/多肽的合成后再修饰对研究翻译后修饰、监测体内的蛋白分布及评估抗体类药物治疗特性等研究至关重要.由于富含电子及在蛋白表面丰度低的特点,酪氨酸(Tyr)通常作为修饰研究的靶点.利用产物捕获的筛选策略,Silverman等[29]分离出可在多肽链中的酪氨酸残基上引入叠氮化修饰的DNA酶[图2(C)].该酶首先催化酪氨酸侧链与2′-叠氮基-dATP之间的反应,形成磷酸二酯键;随后,利用铜催化的叠氮化物-炔烃环加成反应(CuAAC)可将特定的修饰物聚乙烯醇[(PEG)或荧光标签等]附加至叠氮基上,实现对酪氨酸的可检测/可视化标记.筛选获得的DNA酶DzAz2对YPR多肽序列具有选择性,而DzAz8则无序列选择性,可催化游离多肽上的酪氨酸叠氮化修饰.
高效催化蛋白质中酰胺键的水解可为蛋白质组学的研究提供有力的工具.早在2009年,Silverman等[30]就开始尝试寻找可以水解酰胺键的DNA酶,但结果却不尽如人意.基于此,Silverman等[31]对核苷酸进行蛋白质样功能团修饰并利用产物捕获策略分离有催化活性的DNA序列.对文库随机序列中的胸腺嘧啶核苷进行修饰,使其携带氨基、羧基或羟基,以此模拟能水解蛋白质酰胺键的天然蛋白酶;同时,利用5′-氨基寡核苷酸与催化酰胺键水解后的产物DNA序列发生共价交联反应,将其捕获分离[图2(D)].经过如此改进后,他们分离出了多个可催化酰胺键水解的DNA酶,使反应速率提升了3个数量级.
综上可见,近年来诞生的产物捕获筛选策略丰富了DNA酶催化反应的种类,多种可催化蛋白质/多肽生化反应的DNA酶的发现为围绕蛋白质的化学生物学的研究提供了潜在的分子工具.

Fig.2 The product⁃capturing strategy to isolate deoxyribozymes that catalyze reactions on proteins/peptides
1.2 依托环化酶、正负双向及人工核酸的筛选策略
作为遗传信息的携带者,DNA是极其稳定的.从催化的角度来看,水解DNA的磷酸二酯键极具挑战性;但从DNA编辑的角度,水解切割DNA的需求又非常高.一直以来,水解切割DNA是蛋白酶的专利,直到2009年Silverman等[30]发现DNA酶同样可以水解切割DNA,才打破了蛋白酶的垄断.
最近,本课题组[32]提出了一种基于环化酶(Circ Ligase)的高效筛选策略[图3(A)],根据该策略,分离出多个可特异水解切割DNA的DNA酶,它们在体外的反应速率可与蛋白酶相媲美.与其它固定切割位点的筛选策略不同,该方法允许DNA序列在DNA骨架上的任意位置发生水解断裂,主要包括以下步骤:(1)通过搭桥延伸法构建了包含5′-磷酸基团和2个50 nt随机序列的线性单链DNA文库(较长的序列可以赋予文库更多的DNA二级结构);(2)使用环化酶将线性文库连接成环,并用变性聚丙烯酰胺凝胶(PAGE)胶分离环形连接产物,进一步用哌啶处理环化DNA,去除因DNA损伤发生的自切割DNA序列,降低筛选过程中的背景信号;(3)将环状DNA文库在含辅助因子(Zn2+)的筛选缓冲液中孵育,并用变性PAGE胶分离出因切割导致断裂的线性DNA;(4)再次使用环化酶将线性DNA切割产物连接成环(环化酶只能识别并环化连接含5′-磷酸基团和3′-羟基的DNA序列,因此只有水解切割磷酸二酯键的DNA序列能被连接成环,而因氧化损伤等导致断裂成线性的DNA序列则无法成环;此外,环化酶催化的DNA连接无需搭桥序列的帮助,是在切割位点未预先设定的DNA底物被切割后能再次被连接成环的关键);(5)通过PCR扩增上一步中的环形DNA序列,为建立次级文库作准备.经过上述步骤的多轮重复筛选,最终分离出2类可感应Zn2+并快速水解DNA的DNA酶,其中I类酶的催化核心区域仅为15个保守的碱基,在体外的反应速率(kobs)可达到1~3 min-1.目前,这些酶已被用于单链DNA ladder的开发[33]和单链DNA序列的生物法制备[34].

Fig.3 Strategies to isolate deoxyribozymes that catalyze reactions on nucleic acids
N6-腺苷酸甲基化(m6A)是生物体内最广泛的RNA修饰之一,准确区分特定RNA位点的甲基化修饰意义重大.2018年,Sednev等[35]报道了一类可特异识别m6A修饰并切割RNA的DNA酶,该DNA酶具有检测特定RNA序列中的甲基化率的潜力.为了获得识别m6A修饰的特异性,他们采用了双向筛选策略[图3(B)],以含有m6A修饰的RNA(R1)为底物进行正向筛选,以无修饰的RNA(R2)为对照进行负向筛选,通过消除识别未修饰RNA并切割的DNA序列来保证筛选的特异性{[图3(B)]中红色顺时针方向};同时,如果以R2为底物进行正向筛选,以R1为对照进行负向筛选,则可获得特异识别未被m6A修饰的RNA并将其切割的DNA酶{[图3(B)]中绿色逆时针方向}.理论上,这种双向的筛选策略可被进一步推广,用于开发可特异识别转录组中其它RNA修饰的DNA酶.
除了常规DNA外,人工合成核酸(XNA)也具有催化活性[36,37].Holliger等[38]率先提出X-SELEX策略,即指数放大的交叉化学选择性富集技术(Cross-chemistry Selective Enrichment by Exponential Amplification),用于开发人工合成核酸酶(XNAzymes)[图3(C)].X-SELEX的第一步是构建XNA文库(约1014个不同分子),以含有随机序列的DNA文库为模板,同时杂交偶联短的DNA,RNA或XNA,在XNA聚合酶作用下延伸出与DNA模板互补的XNA序列(DNA/XNA双链形式);第二步,利用生物素/亲和素强相互作用,分离获得单链的XNA文库;第三步,将XNA文库置于筛选环境中一段时间后,通过变性PAGE胶回收有催化活性的XNA序列;第四步,利用反转录酶获得与XNA序列互补的互补脱氧核糖核酸(cDNA);第五步,以cDNA为模板,PCR扩增放大信号成次级DNA文库,为下一轮筛选作好准备.采用X-SELEX筛选策略,Taylor等[39]发现了具有RNA内切酶、连接酶和XNA-XNA连接酶活性的人工合成核酸酶;Chaput等[40,41]分离出具有切割RNA活性的核酸酶.X-SELEX筛选策略极大地丰富了人工合成核酸酶的种类.由于XNA不能很好地被天然的脱氧核糖核酸酶(DNase)识别,在生物环境中具备更好的稳定性,因此可能在潜在的体内应用中拥有一定的优势.
2 切割核酸的工具型DNA酶传感器
2.1 特定金属离子作为配体
自DNA酶问世以来,在二十多年的发展历史中,催化切割RNA的DNA酶(RNA-cleaving deoxyribozymes)一直占据重要地位,是研究最多的一类DNA酶.DNA酶催化RNA的切割需要辅因子(特别是金属离子)的参与,一系列响应不同金属离子(主要是二价态)或小分子的切割RNA的DNA酶[42,43],包括响应 Pb2+[44],Ca2+[45],Cd2+[46],Zn2+[47],Fe2+[48],UO22+[49]及镧系金属[50~52]等,已被分离出来. 近年来,随着筛选策略的不断优化,一些新的响应不同价态金属离子的切割RNA的DNA酶陆续被发现.
在筛选过程中,增加对有催化活性DNA序列的分离纯化步骤,即多步分离,可提高筛选效率,缩短筛选轮数.通常,对切割RNA的DNA酶的辅因子选择多为二价金属离子.而一价金属离子(如Na+),由于其中和DNA骨架上负电荷的能力远小于二价金属离子如Mg2+,因此较难辅助DNA序列折叠成具有催化活性的二级结构,这对体外筛选响应一价金属离子的DNA酶探针提出了挑战.2015年,Lu等[53]采用两步分离纯化策略,筛选出了Na+依赖的RNA切割DNA酶探针.先将响应Na+并发生切割RNA底物的DNA序列从固定相上洗脱下来,再用变性PAGE胶对切割下来的DNA信号进行二次纯化分离[图4(A)].在起始核酸文库中,有催化活性的DNA序列比例远远小于无活性的DNA序列.因此,增加的胶纯化步骤可以有效去除从固相柱上非特异性洗脱下来的、未切割的DNA序列,从而降低背景噪音、提高筛选效率.筛选出的DNA酶探针NaA43ES展现出高于其它离子104倍的选择性和良好的检测灵敏度(检测限为135 μmol/L),为细胞内Na+的监测成像提供了新方法.Liu等[54]采用类似方法开发了可以在有机相(乙醇和二甲基亚砜)中感应Na+并切割RNA的DNA酶EtNa.这一系列的研究工作大大拓展了DNA酶的应用范围.
将DNA酶中的保守序列部分进行部分退化后再筛选,可进化出更特异、更活泼的突变体.Ag10C是一类采用经典的筛选策略获得的响应Ag+并切割RNA的DNA酶[55],其催化速率为0.41 min-1.对Ag10C的催化原理进行深入研究后发现,Ag10C可以同时结合2个Ag+,且其对Ag+的选择性不是很高,当Na+存在且浓度升高时可显著提高Ag10C的催化活性[56].在对Ag10C保守序列区域的19个碱基实施部分退化(单个嘌呤由100%退化成50%的鸟嘌呤和50%的腺嘌呤,单个嘧啶由100%退化成50%的胸腺嘧啶和50%的胞嘧啶)并进行重筛选后[图4(B)],获得的突变体AgB1具有较小的催化结构域;与Ag10C相比,AgB1的催化活性提高了200%,并且对Ag+展现出更显著的选择性[57].

Fig.4 Optimizing strategies to select the metal⁃ion⁃dependent RNA⁃cleaving deoxyribozymes
在核酸文库中引入修饰的核苷酸序列可增加筛选的成功率和筛选出的DNA酶的化学多样性,这一策略在筛选切割RNA的DNA酶中多次被使用.在切割位点rA碱基处引入硫代修饰,有利于将亲硫的金属离子招募到rA的磷酸二酯键附近,提高筛选成功率[图4(C)].基于该方案已分离出特异响应Cu2+并切割RNA的DNA酶PSCu10[58],该DNA酶的催化区域只含有8个碱基,切割速率可达0.1 min-1.为了避免筛选到Ce13d这类特异性差的核酸酶(一类可被包括Cu2+在内所有亲硫的金属离子激活的DNA酶),在每一轮的筛选步骤中可通过添加与Ce13d保守区域互补杂交的DNA片段,封闭掉潜在的Ce13d序列的催化活性,从而将其从文库中去除.采用类似的筛选方法也分离得到了特异响应Cr3+和Cr4+并切割RNA的DNA酶[59].
此外,在RNA碱基切割位点处进行咪唑和羧基修饰,也可以为金属离子提供更多的结合位点[图4(D)].根据该策略分离得到的识别Zn2+的切割RNA的DNA酶可结合多达3个Zn2+,并且对Zn2+的特异感应性随着结合Zn2+数目的增多而增强:结合1,2和3个Zn2+的DNA酶对Co2+的选择性分别提高了20,1000和5000倍[60].
除了在切割位点处引入修饰外,在随机序列区引入含修饰的碱基也是常用的一种手段.进行PCR扩增时可在一定程度上引入8-组胺化脱氧腺苷5′-三磷酸(dAimTP)、5-氨基烯丙基脱氧胞苷5′-三磷酸(dCaaTP)和5-胍基烯丙基脱氧尿嘧啶核苷三磷酸(dUgaTP)等修饰过的碱基,生成的含有多个修饰的文库可用来筛选可降解全RNA序列的DNA酶Dz7-38-32[图4(E)][61],该酶在低镁离子(0.5 mmol/L)生理条件下的催化效率可达106L·mol-1·min-1.
由此可见,通过优化筛选策略,如多步纯化信号DNA序列、对已有的DNA酶退化再进化及引入适当的化学修饰等,可全面提升DNA酶的特异性、灵敏度、活性及催化种类,并为围绕其进行生物传感方面的研究提供必要的支撑.
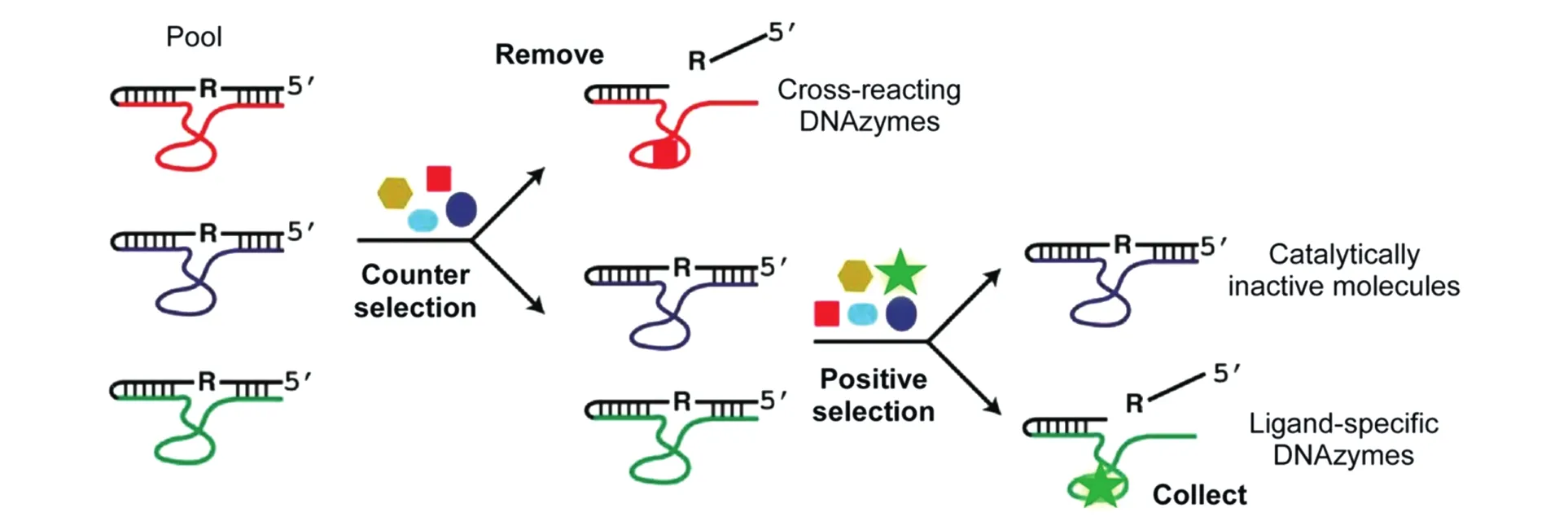
Fig.5 Cancelling strategies for the selection of deoxyribozymes that specifically target a biological sample[64]
2.2 对冲抵消的策略:筛选感应特定生物样本的DNA酶传感器
选择某一特定的、明确的离子或分子作为辅助因子进行RNA切割型的DNA酶的筛选策略已经相对成熟,其中的一部分DNA酶已被进一步编辑,用来作为检测某些生物和化学样本中相应离子或分子的探针.但一些重大恶性疾病的生物样本中(如细菌、细胞的分泌物或胞内提取物等)待检测的目标分子的身份可能是不明确的(如三阴性乳腺癌的亚型繁多,缺乏标志物),对于这类样本能否做到特异性检测是一个极具挑战性的难题.
近十年来,可催化切割RNA的DNA酶作为一个易操作且易编辑的工具,为解决特异识别靶标不明确的生物样本检测的难题提供了高效的解决方案.Li等[62]首创了一种对冲抵消的筛选策略.以细菌为例,根据该筛选策略[63,64],首先各自收集菌株(目标菌株及参照物菌株)的未纯化的粗提代谢混合物(CEM),以CEM为筛选对象确保所筛DNA酶探针在复杂生物样品环境中仍具备催化活性;其次,该筛选策略采用两步筛选法来保证DNA酶探针识别的特异性(图5):(1)负向筛选(Counter selection),即核酸文库与非目标菌株的CEM共孵育,去除发生RNA切割的DNA序列,保留未切割序列进入下一步;(2)正向筛选(Positive selection),分离与目标菌株CEM共孵育后发生切割的DNA序列,利用PCR放大信号进入下一步.这种对冲抵消筛选策略的优势在于无需预先知道靶标分子身份即可获得特异感应目标菌株(代谢物)的DNA酶探针;此外,负向筛选可确保去除目标菌株CEM中的非独有成分对DNA酶切割响应,即通过筛选本身来保证DNA酶探针的特异性.
2011年,Li等[65]采用对冲抵消筛选策略首次获得了特异感应大肠杆菌(E.coliK12)的DNA酶探针RFD-EC1.在筛选过程中,以枯草芽孢杆菌CEM作为对照进行负向筛选,以大肠杆菌CEM(E.coliK12)为研究对象进行正向筛选,从而保证DNA酶探针的特异性.同时,通过在切割位点(rA)两侧的T碱基引入一对荧光基团和猝灭基团,可实现每一轮筛选切割信号的可视化.Li等[66]还采用相同策略获得了特异感应艰难梭状芽胞杆菌BI/027-H菌株的DNA酶探针RFD-CD1;同时,结合分子筛技术和生信分析等手段,与RFD-CD1探针特异相互作用的分子被发现为转录因子TcdC截短突变体.该研究表明,在靶标分子未知情况下,采取对冲抵消筛选策略不仅可以获得特异识别BI/027-H菌株的DNA酶探针,还可以利用所筛探针进一步明确其响应的靶标分子身份.对冲抵消的筛选策略也被用于筛选特异识别水生鳗弧菌CEM的DNA酶VAE-2[67],基于VAE-2再编辑成的荧光生物传感器可检测低至4000 cfu/mL的信号,适用于鱼肉组织和饮水样品中鳗弧菌的检测.
在哺乳类细胞水平上,对冲抵消筛选策略也获得了成功.Li等[68]以正常乳腺细胞MCF-10A的裂解液为对照进行负向筛选,以三阴性乳腺癌细胞MDA-MB-231裂解液为目标进行正向筛选,获得了特异响应MDA-MB-231细胞裂解液并在RNA位点发生切割的DNA酶RFD-AAI2-5.该探针能有效区分MDA-MB-231细胞与正常乳腺细胞及其它亚型乳腺癌细胞,检测限低至约5000 cell/mL.在进一步对反应缓冲液的pH值、辅助因子种类和浓度及DNA序列的有效长度进行优化后,该探针的检测限提高至约2000 cell/mL,且能从3种正常细胞和10种肿瘤细胞中特异识别出MDA-MB-231细胞[69].
除了乳腺癌细胞外,对冲抵消策略也被用于慢性髓细胞性白血病细胞K562的DNA酶探针的筛选[70],分离获得的探针RFD-A1-3可高特异性识别该细胞,围绕此探针可建立一种简单快速、用于早期临床诊断分析慢性髓细胞性白血病的方法.该方法可有效区分K562胞外分泌物与其它白血病亚型细胞、肿瘤细胞系,检测限低至10 nmol/L,且适用于含人血清的复杂环境.
上述研究表明,在靶标未知情况下,利用对冲抵消策略可筛选获得特异识别生物样本(如细菌和细胞等)并在RNA位点发生切割的DNA酶探针,其易于与化学发光相结合及放大检测信号的特点有望为致病菌和重大恶性肿瘤的临床诊断提供便利和解决方案.
2.3 DNA酶在活细胞中的检测和智能化应用
除了Lu等[53]开发的感应Na+的DNA酶探针用于细胞内Na+的监测成像外,近年来,已有多个DNA酶探针被开发并应用于活细胞内特定信号的检测放大和活细胞内药物分子的智能释放.由于DNA酶探针极强的可编辑及可修饰性,它们已被证明可非常方便地与纳米颗粒融合在一起,从而提升被特定细胞摄取的效率.2017年,Zhao等[71,72]报道了可利用氧化锰(MnO2)纳米纸片携带感应锰离子(Mn2+)的DNA酶探针在活细胞内监测放大DNA碱基缺失修复的信号和活细胞中端粒酶的信号.MnO2纳米纸片不仅作为DNA酶探针的载体,在进入活细胞后,其可被还原成Mn2+,从而触发DNA酶探针的切割;待检测的细胞内信号分子,如DNA碱基缺失修复酶和端粒酶等,被设计成与MnO2还原为Mn2+直接关联,因此DNA酶探针在活细胞内的切割信号就可以反映待检测的胞内分子的水平.2019年,Wang等[73]报道了利用氧化锌(ZnO)纳米颗粒携带感应锌离子Zn2+的DNA酶探针和多柔比星(Dox)药物分子用于对癌细胞智能化的释放药物.DNA酶在活细胞中的检测和智能化应用刚刚开端,极有可能是下一个5~10年的研究热点.
3 总结与展望
近年来,针对DNA酶的筛选策略不断发展和完善,新发现的DNA酶的种类越来越丰富,特异性和灵敏度都在提升,这也促进了DNA酶作为一种生物型检测工具的应用.但DNA酶的筛选过程仍然比较繁琐且耗时,如何借助微流控等微纳技术实现DNA酶的高通量和自动化筛选,如何缩短开发DNA酶的时间和人力成本,是未来需要解决的问题.目前,DNA酶已在生物传感和基因治疗等领域崭露头角,未来也很可能会进一步在生物医疗领域大放异彩.在临床上发挥作用之前,DNA酶还需突破包括器官、组织和细胞等在内的重重障碍;如何有效递送DNA酶并使之在体内发挥等同于体外的功能,并能同时抵抗生理环境中DNase的降解,是推进DNA酶应用需解决的另一问题.解析DNA酶的晶体结构有助于科学家们更好地理解其功能,并设计合理的筛选策略开发新的DNA酶.当前,已有2个DNA酶的晶体结构被成功解析[14,15].伴随着结构生物学的迅猛发展,相信越来越多的DNA酶将会在原子/分子水平上得到解析,人类对于DNA酶的了解、掌握和应用必将越来越深入.
猜你喜欢
猪业科学(2021年3期)2021-05-21
云南化工(2020年11期)2021-01-14
教学考试(高考生物)(2020年6期)2020-11-23
幽默大师(2020年10期)2020-11-10
矿产综合利用(2020年1期)2020-07-24
食品与生物技术学报(2020年8期)2020-01-06
中华诗词(2019年1期)2019-11-14
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
猪业科学(2018年4期)2018-05-19
