
一种光酸响应型[2] 轮烷分子梭的合成与表征
2020-11-06 08:30曲大辉
华东理工大学学报(自然科学版) 2020年5期
茆 敏, 刘 月, 杨 舜, 曲大辉
(华东理工大学化学与分子工程学院,结构可控先进功能材料及其制备教育部重点实验室,上海 200237)
自从Stoddart 等[1]创造了首个分子梭后,人造分子机器迎来了巨大的发展机遇。由于刺激响应型的分子梭在机械互锁的纳米体系中具有独特的穿梭运动能力,其在分子机器[2-5]、自修复材料[6-8]和可响应的超分子聚合物[9-11]的构建过程中具有非常广泛的应用。这些功能和应用大多基于双稳态的轮烷型分子梭,主要的调控策略包括酸碱、光热和氧化还原刺激[12-14]。但是,如何在复杂的体系中选择合适且高效的调控手段一直是亟待解决的难题。
光化学刺激被认为是调控分子机器的最有前景、最具应用价值的模式,因为光控不仅在时间和空间上具有精确性,而且具有简便、清洁、无污染等优点[15]。光控轮烷型分子机器大多通过在轮烷骨架上引入偶氮苯或者二苯乙烯等光致异构化组件实现光控性能[16]。Harada 等[17]利用α-环糊精和偶氮苯衍生物上的相互作用形成以[2] 轮烷单元为聚合物主链的拓扑交联,制备了一种水性光响应聚合物材料。但是在复杂的人造分子机器中,这类光致异构化基团通常难以完全实现异构化[18]。除了利用光致异构分子外,本课题组利用“光扳机”的策略调控轮烷型分子梭,虽然该方法转化率高,但是既产生废物又不是可逆过程[19]。
光致酸产生剂(光酸)是一种在光照下发生反应或解离的化合物,在这种反应或解离中,最终形成的光产物包括溶液或固体中的酸性物质。传统的光酸化合物可分为两大类:离子型和非离子型[20]。与传统的利用自由基、阳离子或分解过程不同的是,许多化学家提出了“光引发-酸生酸”的替代方法,最常见的新型光酸系统是部花青(MEH)和螺吡喃(SP)的互变体系[21]。Liao 等[22]研究了在光照下,一系列部花青化合物的pH 如何从弱酸性(5.5)转移到强酸性(3.2),因此认为它们可以用作氢的来源,催化费歇尔酯化反应以及改变pH 敏感聚合物的状态。基于这类分子优异的光控释酸的性质,Silvi 等[23]通过光控的部花青和螺吡喃光酸系统可逆控制拟轮烷的络合和解离。因此在酸碱响应型主客体系统中,光诱导质子转移机制是一种可利用光化学刺激间接可逆实现酸调控的有效策略。
目前为止,有关光酸对轮烷型分子梭调控作用的系统性的研究报道较少。本文设计和构建了利用光致酸产生剂部花青调控[2] 轮烷的梭动过程。部花青和螺吡喃在互变过程中可释放和吸收质子,以此来控制[2] 轮烷R 分子梭“运动”和“静止”状态的多次可逆切换。利用核磁共振氢谱(1H-NMR)、核磁共振碳谱(13C-NMR)和高分辨质谱(HRMS)等手段确定轮烷分子梭结构的正确性,并通过变温核磁共振氢谱计算分子梭中的联吡啶嗡环(CBPQT4+)在杆上的穿梭速率。
1 实验部分
1.1 仪器和试剂
1H-NMR 的测定采用Bruker AM 400(400 MHz)和Bruker Ascend-600(600 MHz)核磁共振波谱仪进行表征,以四甲基硅烷(TMS)作为内标。13C-NMR由Bruker AM 400(100 MHz)核磁共振波谱仪进行表征。电喷雾质谱法(ESI-MS)用Waters LCT Premier XE 质谱仪测定。紫外-可见吸收光谱使用Shimadzu UV-1700/UV-3600 紫外光谱仪测试。
所有实验操作未经特殊说明,均在惰性气体(N2,Ar)的保护下以史莱克(Schlenk)无水、无氧操作技术的标准进行。实验过程中所使用的溶剂和试剂均为市售分析纯或化学纯,除特别注明外,所有溶剂和试剂均未经实验室进一步提纯处理。相关中间体5[24], 7[25]分别根据有关参考文献方法进行合成。
1.2 [2] 轮烷R 的合成路线
[2] 轮烷R 的合成路线及化合物1~7 的化学结构式如图1 所示。
1.2.1 化合物1 的合成 500 mL 单口茄形烧瓶中加入四甘醇(50 g, 257 mmol)和三乙胺(6.2 g, 61.7 mmol),再加入300 mL 二氯甲烷(CH2Cl2)溶剂进行溶解,将反应容器置于0~5 ℃冰水浴下,用恒压滴液漏斗缓慢滴加50 mL 对甲苯磺酰氯(TsCl, 9.8 g, 51.4 mmol)的CH2Cl2溶液,滴毕撤去冰浴,将反应液置于室温下搅拌3 h,薄层层析色谱法跟踪反应,直到化合物TsCl反应完毕。反应结束后,用饱和食盐水(3×100 mL)洗涤,然后合并有机相,无水硫酸钠(Na2SO4)干燥30 min,抽滤,取滤液旋干,得到粗产品无色油状黏稠液体。粗产品采用硅胶柱层析(硅胶颗粒为200~300 目(48~75 μm),CH2Cl2)的方法进行纯化,得到无色油状液体化合物1 13.4 g,产率75%。1H-NMR(400 MHz, CDCl3, δ) 7.83 (d, J=8.5 Hz, 2H), 7.37 (d,J=8.5 Hz, 2H), 4.19 (t, J=8.5 Hz, 2H), 3.6~3.8 (m, 14H),2.48 (s, 3H)。13C-NMR (100 MHz, CDCl3,δ) 143.6,131.9, 130.3, 128.4, 83.7, 72.9, 71.1, 70.9, 70.7, 69.7,69.1, 62.2, 22.1。 HRMS (ESI) (m/z): [M+Na]+calcd for C15H24SO7Na: 371.113 5, found: 371.114 0。
1.2.2 化合物2 的合成 250 mL 单口茄形烧瓶中加入化合物1(10 g, 28.7 mmol)、1,5-二羟基萘(2.1 g,13.0 mmol)和碳酸钾(5.4 g, 39.0 mmol),再加入100 mL N,N-二甲基甲酰胺(DMF)溶剂进行溶解,将反应容器加热至90 ℃,氩气氛围下反应24 h。薄层层析色谱法跟踪反应,直到化合物1,5-二羟基萘反应完毕。反应结束后,反应液冷却到室温,然后抽滤除去碳酸钾,取滤液旋干,加入50 mL CH2Cl2溶解,用饱和食盐水(3×50 mL)洗涤,然后合并有机相,无水Na2SO4干燥30 min,抽滤,取滤液旋干,得到粗产品。粗产品采用硅胶柱层析(硅胶颗粒为200~300目(48~75 μm),CH2Cl2与CH3OH 体积比30∶1)的方法进行纯化,得到化合物2 4.0 g,产率60%。1H-NMR(400 MHz, CDCl3,δ) 7.87 (d, J=8.0 Hz, 2H), 7.35 (t,J=8.0 Hz, 2H), 6.85 (d, J=8.0 Hz, 2H), 4.27~4.33 (m,4H), 3.96~4.02 (m, 4H), 3.78~3.84 (m, 4H), 3.55~3.73(m, 20H)。13C-NMR (100 MHz, CDCl3,δ) 154.4, 126.8,125.1, 114.6, 105.8, 72.5, 71.0, 70.7, 70.3, 69.8, 67.9,61.7。 HRMS (ESI) (m/z): [M+H]+calcd for C26H41O10:513.269 4, found: 513.269 9。
1.2.3 化合物3 的合成 250 mL 单口茄形烧瓶中加入化合物2(4.0 g, 7.8 mmol)和三乙胺(0.78 g, 7.8 mmol),再加入100 mL CH2Cl2溶剂进行溶解,将反应容器置于0~5 ℃冰水浴中,用恒压滴液漏斗缓慢滴加30 mL TsCl (1.0 g, 5.2 mmol)的二氯甲烷溶液,滴毕撤去冰浴,将反应液置于室温下搅拌12 h,薄层层析色谱法跟踪反应,直到化合物TsCl 反应完毕。反应结束后,用饱和食盐水(3×100 mL)洗涤,然后合并有机相,无水Na2SO4干燥30 min,抽滤,取滤液旋干,得到粗产品无色油状黏稠液体。粗产品采用硅胶柱层析(硅胶颗粒为200~300 目(48~75 μm),CH2Cl2与CH3OH 体积比100∶1)的方法进行纯化,得到化合物3 1.75 g,产 率50%。1H-NMR (400 MHz, CDCl3,δ) 7.75 (d,J=8.4 Hz, 1H), 7.74 (d, J=8.4 Hz, 1H), 7.61 (d, J=8.0 Hz,2H), 7.21 (t, J=8.4 Hz, 2H), 7.12 (d, J=8.0 Hz, 2H), 6.68(d, J=8.4 Hz, 2H), 4.07~4.11 (m, 4H), 3.94~3.97 (m,2H), 3.77~3.81 (m, 4H), 3.58~3.62 (m, 4H), 3.51~3.55(m, 4H), 3.46~3.50 (m, 8H), 3.39~3.41 (m, 4H),3.35~3.37 (m, 2H), 2.2 (s, 3H)。13C-NMR (100 MHz,CDCl3, δ ) 154.2, 144.7, 132.8, 129.8, 127.7, 126.6,125.1, 114.5, 105.6, 72.4, 70.7, 70.5, 70.4, 70.4, 70.4,70.3, 70.1, 69.6, 69.3, 68.4, 67.8, 61.4, 21.4。 HRMS(ESI) (m/z): [M+H]+calcd for C33H47O10S: 667.278 3,found: 667.278 0。
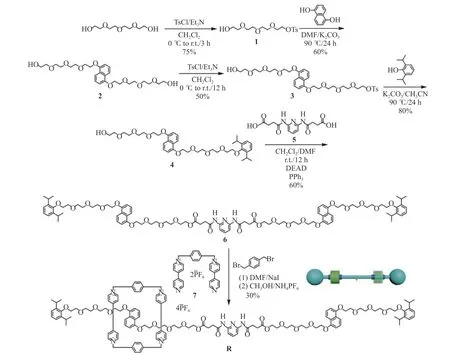
图 1 [2] 轮烷R 的合成路线Fig. 1 Synthetic route of [2]rotaxane R
1.2.4 化合物4 的合成 250 mL 单口茄形烧瓶中加入化合物3(1.75g, 2.6 mmol)、2,6-二异丙基苯酚(0.7 g,3.9 mmol)和碳酸钾(0.72 g, 5.2 mmol),再加入100 mL乙腈溶剂进行溶解,将反应容器加热至90 ℃回流,氩气氛围下反应24 h。薄层层析色谱法跟踪反应,直到化合物3 反应完毕。反应结束后,反应液冷却到室温,然后抽滤除去碳酸钾,滤液旋干后加入50 mL CH2Cl2溶解,用饱和食盐水(3×50 mL)洗涤,然后合并有机相,无水Na2SO4干燥30 min,抽滤,取滤液旋干,得到粗产品。粗产品采用硅胶柱层析(硅胶颗粒为200~300 目(48~75 μm),CH2Cl2与CH3OH 体积比120∶1)的方法进行纯化,得到化合物4 1.4 g,产率80%。1H-NMR (400 MHz, CDCl3,δ) 7.87 (d, J=7.8 Hz,2H), 7.34 (t, J=7.8 Hz, 2H), 7.09 (s, 3H), 6.84 (d, J=7.8 Hz, 2H), 4.31 (t, J=4.9 Hz, 4H), 3.68~4.02 (m, 26H),3.59 (t, J=4.6 Hz, 2H), 3.39 (m, 2H), 1.22 (d, J=6.9 Hz,12H)。13C-NMR (100 MHz, CDCl3,δ) 154.6, 153.3,142.1, 127.0, 125.4, 124.9, 114.9, 105.9, 74.1, 72.8, 71.3,71.27, 71.2, 71.1, 71.04, 71.0, 70.9, 70.8, 70.6, 70.1,68.2, 68.1, 61.9, 26.5, 24.4。 HRMS (ESI) (m/z):[M+H]+calcd for C38H57O10: 673.394 6, found: 673.394 2。1.2.5 化合物6 的合成 100 mL 单口茄形烧瓶中加入化合物4(1.4 g, 2.1 mmol)、化合物5(325 mg, 1.05 mmol)、偶氮二甲酸二乙酯(DEAD, 402 mg, 2.31 mmol)和三苯基膦(PPh3, 606 mg, 2.31 mmol),再加入45 mL CH2Cl2和9 mL DMF 的混合溶剂进行溶解,将反应容器用铝箔纸包住避光,氩气氛围下常温反应12 h。薄层层析色谱法跟踪反应,直到化合物4 反应完毕。反应结束后,旋干反应液中的二氯甲烷后加入50 mL乙酸乙酯溶解,用饱和食盐水(3×90 mL)洗涤,然后合并有机相,无水Na2SO4干燥30 min,抽滤,取滤液旋干,得到粗产品。粗产品采用硅胶柱层析(硅胶颗粒为200~300 目(48~75 μm),CH2Cl2与CH3OH 体积比100∶1)的方法进行纯化,得到化合物6 1.0 g,产率60%。1H-NMR (400 MHz, DMSO-d6,δ) 10.14 (s, 2H),7.69~7.74 (m, 7H), 7.34~7.39 (m, 4H), 7.06~7.10 (m,6H), 6.97 (d, J=8.0 Hz, 4H), 4.22~4.26 (m, 8H),4.09~4.11 (m, 4H), 3.87~3.90 (m, 8H), 3.78~3.80 (m,4H), 3.55~3.72 (m, 32H), 3.49~3.53 (m, 8H), 2.59~2.70(m, 8H), 1.14 (d, J=4.0 Hz, 24H)。13C-NMR (100 MHz,DMSO-d6,δ) 173.7, 173.1, 153.6, 149.6, 148.7, 141.5,139.9, 128.2, 125.2, 123.9, 123.7, 115.6, 111.7, 107.6,74.2, 70.4, 70.1, 66.9, 32.1, 28.9, 27.6, 23.6。 HRMS(ESI) (m/z): [M+Na]+calcd for C89H123N3O24Na:1 640.838 9, found: 1 640.869 3。
1.2.6 目标化合物[2] 轮烷R 的合成 50 mL 单口茄形烧瓶中加入化合物6(1.0 g, 0.62 mmol)、化合物7(220 mg, 0.31 mmol)、1,4-二(溴甲基)苯(82 mg,0.31 mmol)和碘化钠(5 mg, 0.031 mmol),再加入15 mL DMF 进行溶解,氩气氛围下常温反应5 d。薄层层析色谱法跟踪反应,直到化合物7 反应完毕。反应结束后,旋干DMF 后加入50 mL 甲醇溶解,然后再加入10 mL 饱和六氟磷酸铵水溶液进行离子交换,常温搅拌过夜。反应结束后用CH2Cl2(3×60 mL)萃取,合并有机相,无水Na2SO4干燥30 min,抽滤,取滤液旋干,得到粗产品。粗产品采用硅胶柱层析(硅胶颗粒为200~300 目(48~75 μm),CH2Cl2与CH3OH 体 积 比10∶1)的方法进行纯化,得到紫色的目标化合物[2] 轮烷R 254 mg,产率30%。1H-NMR (400 MHz,CD3CN,δ) 10.01 (d, J=16.0 Hz, 2H), 8.81 (m, 2H),7.91~8.03 (m, 9H), 7.59~7.62 (m, 2H), 7.18~7.34 (m,9H), 7.02~7.12 (m, 6H), 6.82 (t, J=8.0 Hz, 2H),6.71~6.74 (m, 2H), 6.24~6.27 (m, 2H), 6.03 (s, 4H),5.94~5.99 (m, 2H), 5.89 (s, 4H), 5.76 (s, 8H), 4.30~4.31(m, 4H), 4.20~4.24 (m, 8H), 4.12~4.14 (m, 2H),4.03~4.09 (m, 8H), 3.95~3.99 (m, 6H), 3.90~3.92 (m,2H), 3.77~3.81 (m, 6H), 3.65~3.72 (m, 16H), 3.54~3.58(m, 10H), 3.45~3.47 (m, 2H), 3.30~3.37 (m, 4H),2.73~2.76 (m, 4H), 2.58~2.65 (m, 4H), 2.42 (d, J=8.0 Hz, 2H), 1.11~1.17 (m, 24H)。13C-NMR (100 MHz,DMSO-d6,δ) 173.7, 173.1, 153.6, 150.7, 149.6, 148.7,144.6, 141.5, 139.9, 131.4, 128.9, 128.2, 126.5, 125.2,123.9, 123.7, 115.6, 111.7, 107.6, 74.2, 70.4, 70.1, 66.9,59.6, 32.1, 28.9, 27.6, 23.6。 HRMS (ESI) (m/z): [M-3PF6]3+calcd for C125H155PF6N7O24: 761.359 4, found:761.359 0。
2 结果与讨论
2.1 [2] 轮烷R 的核磁共振氢谱表征
采用核磁共振氢谱对目标化合物[2] 轮烷R 的分子结构进行了分析确认,如图2 所示。化学位移10.01 处的双峰为酰胺键氮原子上的质子峰,8.81 处的双峰是CBPQT4+大环吡啶上邻近氮原子的质子峰,7.59~7.62 处 的 多 重 峰、6.82 处 的 三 重 峰 和6.71~6.74 处的多重峰是未被CBPQT4+大环络合的萘环上的质子峰,6.24~6.27 处的多重峰、5.94~5.99 处的多重峰和2.42 处的双峰是被CBPQT4+大环络合的萘环上的质子峰,1.11~1.17 处的多重峰是封堵基团上甲基上的质子峰。由于CBPQT4+大环与萘环的静电作用,被CBPQT4+大环络合的萘环上质子峰的化学位移移向高场,3、4、5 位质子峰的化学位移远大于6、7、8 位质子峰的化学位移。夸张的是,8 位质子峰受到屏蔽作用的影响,化学位移竟然从芳香区移到了2.42。由于CBPQT4+大环络合在一个萘环上,穿梭运动很慢,封堵基团a 上的质子峰由双峰裂分为四组峰。
2.2 部花青与螺吡喃互变过程
部花青和螺吡喃的转换过程的紫外-可见吸收光谱如图3 所示。初始状态下,部花青溶解于乙腈(298 K, 1.0×10−5mol/L),于420 nm 处有一个特征吸收峰,在419 nm 的光照(3×1 min)下,420 nm 处的吸收峰剧烈降低,在300 nm 处产生了一个新的吸收峰。对比螺吡喃的吸收峰可知,300 nm 处的吸收峰是螺吡喃的特征吸收峰,由此可知,在419 nm 的光照下,部花青转换为螺吡喃。然后将此溶液放置在暗处避光10 min,发现300 nm 处的吸收峰消失,420 nm处的特征吸收峰重新出现,由此可知,在暗处放置10 min后,螺吡喃转换为部花青。

图 3 MEH 与SP 互变过程(a)的紫外-可见吸收光谱(b)Fig. 3 UV-Vis absorption spectra (b) of the conversion between MEH and SP(a)
2.3 [2] 轮烷R 的动力学研究
采用一系列变温核磁共振氢谱(CD3CN, 600 MHz,3.1 mmol/L)研究[2] 轮烷 R 的动力学性能,如图4 所示。随着温度的升高,核磁共振氢谱中封堵基团上甲基氢的位移信号峰越来越窄,并且从298 K 时的四组峰逐步变为338 K 时的双峰,根据核磁共振氢谱的线形分析理论(LSA),说明分子由不对称逐步转为对称结构,即CBPQT4+大环在298 K“静止”在分子一端的萘环上,此时穿梭速率为4.24 s−1;温度升高后,CBPQT4+大环逐步在分子两端的两个萘环之间运动,大环穿梭的速率越来越快,在338 K 时,CBPQT4+大环在杆状组分上处于“运动”状态,此时穿梭速率为242.01 s−1。
进一步研究了光酸调控[2] 轮烷R 的穿梭速率。因为在常温(298 K)下,CBPQT4+大环“静止”在分子一端的萘环上,不是运动状态,所以选择在338 K下进行核磁共振氢谱对比。如图5(a)所示,初始状态(338 K)下,CBPQT4+大环在分子两端的两个萘环之间运动,核磁共振氢谱中封堵基团上甲基氢是双峰,加入光酸部花青后,用419 nm 光照30 min,甲基氢由双峰变为四组峰(见图5(b)),它不是完全裂分的四重峰,更像是完全裂分的四重峰和原双峰的叠加,说明部花青转换为螺吡喃并释放氢质子(见图3(a)),氢质子被[2] 轮烷R 上的吡啶捕获形成空间位阻,一部分分子梭中CBPQT4+大环“静止”在分子一端的萘环上,另一部分分子梭仍然保持初态,分子梭中CBPQT4+大环在分子两端的两个萘环之间运动。将溶液放置在暗处12 h 后,a 质子峰由四组峰变为双峰,螺吡喃SP 夺取氢质子转换为部花青,[2] 轮烷R 分子中间的空间位阻消失,CBPQT4+大环在分子两端的两个萘环之间运动,完全处于“运动”状态。这种循环可以重复很多次。

图 5 (a)光酸调控“静止”与“运动”状态过程的示意图;(b)光酸调控“静止”与“运动”状态过程的核磁氢谱对比Fig. 5 (a) Schematic representation of photoacid controlled STOP-GO process; (b) 1H-NMR spectra of photoacid controlled STOP-GO process
3 结束语
采用模板法成功合成了具有光酸响应的[2] 轮烷分子梭R。在初始状态(338 K)下,CBPQT4+大环在分子两端的两个萘环之间运动,分子梭处于“运动”状态;加入光酸(部花青)后,用419 nm 光照,部花青转换为螺吡喃并释放氢质子,氢质子被[2] 轮烷R 上的吡啶捕获形成空间位阻,分子梭中CBPQT4+大环“静止”在杆状分子一端的萘环上。将体系放置在暗处后,螺吡喃夺取氢质子转换为部花青,[2] 轮烷R 分子中间的空间位阻消失,CBPQT4+大环在分子两端的两个萘环之间运动,分子梭恢复“运动”状态。因此,光诱导质子转移是一种高效、绿色的可实现轮烷调控的策略,为设计和应用光响应分子机器提供了新的思路。
猜你喜欢
皮肤病与性病(2021年3期)2021-07-30
科学导报·科学工程与电力(2019年26期)2019-08-13
华东师范大学学报(自然科学版)(2019年2期)2019-06-11
新课程·下旬(2018年10期)2018-01-28
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
中国新闻周刊(2017年7期)2017-03-22
三联生活周刊(2017年9期)2017-03-03
