
免疫相关运动障碍临床研究进展
2020-11-04 12:14石红琴孟环宇梁华峰陈晟
中国现代神经疾病杂志 2020年9期
石红琴 孟环宇 梁华峰 陈晟
运动障碍作为临床常见症候群不仅是神经变性病的特点,同时亦是感染启动免疫机制、神经系统自身免疫性疾病乃至免疫相关治疗引发疾病的重要临床表现。例如,Sydenham 舞蹈病(SC)或人类免疫缺陷病毒(HIV)感染所导致的舞蹈病已为人们所熟识,而由神经系统自身免疫性相关抗体所介导的运动障碍则有多种类型,包括富亮氨酸胶质瘤失活基因1(LGI1)抗体诱发的典型运动障碍面-臂肌张力障碍发作(FBDS),以及谷氨酸脱羧酶(GAD)抗体介导的僵人综合征(SPS)和N-甲基-D-天冬氨酸受体(NMDAR)抗体介导的口-面部运动障碍等。除上述自身免疫性抗体相关运动障碍外,由细胞程序性死亡蛋白1(PD1)抑制剂或嵌合抗原受体T 细胞(CAR-T)疗法等所诱发的运动障碍也逐渐引起关注。就发病机制而言,免疫相关运动障碍包括中枢性和周围性(周围神经和神经肌肉接头)运动障碍两类,其中以中枢神经系统免疫相关运动障碍较为常见。为了使临床医师更好的认识免疫相关性疾病、了解免疫调节治疗与不同类型运动障碍之间的关系,本文对近年免疫相关运动障碍临床研究进展进行归纳阐述,以供临床参考。
一、免疫相关性疾病与运动障碍
1.免疫相关舞蹈病 舞蹈病是免疫相关运动障碍的常见发病形式,其舞蹈样动作可由感染或自身免疫性脑炎等因素所诱发,儿童舞蹈样动作的主要病因是Sydenham 舞蹈病;青年女性主要考虑系统性红斑狼疮(SLE)和抗磷脂综合征(APS)相关舞蹈病,呈亚急性发病,表现为单侧或双侧肢体舞蹈样动作,18F-FDG PET 显像提示纹状体呈高代谢[1]。成年期发病的舞蹈病病因复杂,HIV 感染是感染性免疫相关舞蹈病较为常见的病因,因此对于无运动障碍家族史但疑似感染是致病原因的成年舞蹈病患者,应尽早施行HIV 检测[2]。此外,由自身免疫性相关抗体介导的舞蹈病也是成年免疫相关舞蹈病的重要原因,其中副肿瘤性舞蹈病好发于男性,发病年龄较晚,病程中常伴有周围神经病变和体重减轻[3],除Hu、Yo 抗体等临床广泛关注的副肿瘤相关抗体外,与小细胞肺癌(SCLC)、胸腺瘤、结肠癌、肾癌、淋巴瘤等密切相关的塌陷反应调节蛋白5(CRMP5)抗体也与其舞蹈样动作有关,患者可伴视觉障碍[4]。常见的自身免疫性相关抗体还包括谷氨酸脱羧酶65(GAD65)、接 触 蛋 白 相 关 蛋 白(CASPR2)[3]、IgLon5[5]和LG1 抗 体[6],这些抗体主要与系统性红斑狼疮的发病有关。
2.免疫相关小脑共济失调 是一组由免疫介导的、呈急性或亚急性发病、快速进展的小脑共济失调,包括副肿瘤性小脑变性(PCD)、GAD65 抗体相关性小脑共济失调、麸质共济失调(GA)等。(1)副肿瘤性小脑变性:是副肿瘤综合征的临床常见类型,尤其好发于Yo 抗体阳性的女性病例,可伴发乳腺癌、卵巢癌或其他妇科肿瘤。约有65%的患者在肿瘤确诊之前即已出现躯干和肢体共济失调、构音障碍、眼球震颤等中枢神经系统症状与体征,经针对原发肿瘤治疗后症状可有所改善,但大多数患者预后不良,中位生存期为13 ~22 个月[7];根据文献报道,亦可见于Yo 抗体阳性合并胃肠道腺癌的男性副肿瘤性小脑变性病例[8]。约有32%的副肿瘤性小脑变性患者Hu 抗体阳性,除累及小脑外,还可有边缘系统、脑干、周围神经、自主神经等受累表现,其中65%~75%患者合并小细胞肺癌,后者抗肿瘤治疗和免疫调节治疗效果欠佳,中位生存期仅7 ~13 个月[9]。电压门控性钙离子通道(VGCC)抗体相关副肿瘤性小脑变性仅见于男性,约81%的患者合并小细胞肺癌,44%存在Lambert-Eaton 综合征临床或电生理学表现,免疫调节治疗反应欠佳,中位生存期约12 个月[10];而VGCC 抗体阳性的非肿瘤性小脑变性患者,免疫调节治疗效果良好[11]。约有86%的Ri抗体阳性的副肿瘤性小脑变性患者合并有乳腺癌或肺癌,除表现为小脑共济失调外,分别有36% ~50%的患者出现斜视性眼阵挛、24%出现颌-颈肌张力障碍或喉痉挛,中位生存期约69 个月[12]。此外,还有霍奇金淋巴瘤(HL)相关Tr 抗体、非小细胞肺癌和胸腺瘤相关CRMP5 抗体导致的副肿瘤性小脑变性的个案报告[13]。代谢性谷氨酸受体1(mGluR1)抗体与小脑共济失调也有关,部分患者在其共济失调发生之前即已出现味觉障碍,常伴发霍奇金淋巴瘤,免疫调节治疗效果良好[14]。此外,一些少见抗体 如Homer3、CARP Ⅷ、Sj/ITPR1、GluR δ 2、PKC γ、CA/ARGHAP26、Nb/AP3B2 抗体亦可见于小脑共济失调患者,可合并卵巢癌、乳腺癌、黑色素瘤、胆囊癌、肺癌等[15-17]。(2)GAD65 抗 体 相关 小 脑 共 济失调:以50 ~60 岁女性好发,呈亚急性或慢性发病,临床特点为共济失调与癫发作,并且叠加僵人综合征[18],常合并自身免疫性疾病,如1 型糖尿病、桥本甲状腺炎(HT)等[19],糖皮质激素治疗后血清和脑脊液GAD 抗体水平下降,症状明显改善[20]。(3)麸质共济失调:系一种对谷蛋白过敏的自身免疫性小脑共济失调,常见于40 ~50 岁女性,呈慢性或隐匿性发病,躯干共济失调明显重于肢体,约50%患者可同时存在麸质敏感性肠病或伴局灶性肌阵挛、腭震颤等症状与体征,血清和脑脊液谷氨酰胺转胺酶2 和6(TG2 和TG6)抗体阳性[21]。无麸质饮食是首选治疗方案,其疗效取决于临床症状严重程度或小脑萎缩程度,因此一经确诊应尽早启动无麸质饮食[21]。
3. 免疫相关肌张力障碍以及帕金森综合征
(1)基底节脑炎:临床表现为孤立性皮质下功能异常,尤以运动障碍如肌张力障碍、帕金森样症状或舞蹈样动作为主,同时可以伴有嗜睡和精神症状。头部MRI 基底节区T2WI 呈高信号,随访期间可见基底节区萎缩和胶质增生。基底节脑炎与多巴胺D2 受体(D2R)抗体阳性密切相关,若于疾病早期施行免疫调节治疗,可有效改善临床症状并使D2R抗体水平下降[22],但仅行糖皮质激素治疗而未采取与免疫调节药物联合治疗的患者则残留神经精神后遗症且D2R 抗体可在很长一段时间内持续位于较高水平[23]。(2)面-臂肌张力障碍发作:是LGI1 抗体脑炎的特征性发作症状,表现为单侧面部、手臂频繁而短暂的肌张力障碍样不自主动作,亦可累及下肢或躯干,每次发作持续数秒,每日发作数次至数十次。有学者将面-臂肌张力障碍发作归为癫发作,但大部分患者发作期视频脑电图检测不到节律性变化或样放电[24],因此有学者将其定位于皮质下区域[25]。Flanagan 等[26]发现,面-臂肌张力障碍发作患者头部MRI 可见发作对侧基底节区异常信号,因此更倾向于该病是肌张力障碍而非癫发作。(3)抗NMDAR 脑炎:除精神行为异常、癫发作外,可伴发多种运动障碍,如肌张力障碍、帕金森综合征等,其中以口-面部运动障碍最具特征性,表现为噘嘴、咀嚼、重复做鬼脸样动作。抗NMDAR 脑炎好发于青年女性,易罹患畸胎瘤,也可继发于中枢神经系统感染启动的免疫反应,单纯疱疹病毒(HSV)为其常见病原体,其次为流行性乙型脑炎病毒、EB 病毒、水痘-带状疱疹病毒(VZV)、人疱疹病毒6 型等[27-29]。若于单纯疱疹病毒性脑炎(HSE)后出现运动障碍,更加提示单纯疱疹病毒诱发抗NMDAR 脑炎,而非单纯疱疹病毒性脑炎复发[29]。(4)副肿瘤性颌肌张力障碍和喉痉挛:是Ri 抗体相关副肿瘤综合征的常见伴随症状,发生率约24%,好发于女性,大多数患者合并乳腺癌或肺癌,病情严重者甚至可出现进食困难[12]。(5)副肿瘤性帕金森综合征:主要表现为运动迟缓、姿势不稳、肌强直、眼球运动障碍等,对左旋多巴治疗反应欠佳,常见于Ma1/Ma2 抗体阳性的边缘性脑炎,除帕金森综合征外,还可表现为癫发作、快速眼动睡眠期行为障碍(RBD)、发作性睡病、构音障碍、猝倒发作等,MRI 呈现高位脑干,以及丘脑异常信号[30]。Ma2 抗体阳性常见于睾丸肿瘤患者;而Ma1 抗体则高表达于肺癌,其次是睾丸肿瘤,此类患者神经功能预后不良,对免疫调节治疗反应较差或完全无反应[31]。Kelch 样蛋白11(KLHL11)抗体与精原细胞瘤密切相关,KLHL11 抗体阳性的边缘性脑炎主要表现为共济失调和复视,与Ma2 抗体阳性的边缘性脑炎患者不同,KLHL11 抗体阳性患者对免疫治疗反应良好[32]。此外,亦有文献报道LGI1 和CASPR2 抗体可导致帕金森综合征[33-34]。2006 年,梅奥诊所曾报告4 例GAD65 抗体阳性的帕金森综合征病例,均呈现运动迟缓、垂直性核上性眼肌麻痹表现,此类患者更好发于非裔美国人,免疫调节治疗对病情有所改善[35]。(6)HIV 相关帕金森综合征:发生率约5%,表现为帕金森综合征的HIV 阳性患者均存在严重免疫抑制现象,外周血CD4+T 细胞数目明显减少[36]。2001 年,Hersh 等[37]曾 报 告1 例HIV 阳 性 的 帕 金 森综合征患者采用高效抗逆转录病毒疗法(HAART)治疗后,外周血CD4+T 细胞数目恢复正常,病情明显改善。
4.僵人综合征 由梅奥诊所1956 年首次报告,是临床罕见的神经系统免疫性疾病,以躯干中轴肌和四肢肌僵硬、痛性肌肉痉挛为特征,常伴发自主神经功能障碍如大量出汗、心动过速等症状,女性发病率约为男性的5 倍[38]。该综合征分为经典型和变异型两种类型,后者包括僵肢综合征(SLS)、伴强直和肌阵挛的进展性脑脊髓炎(PERM)[38]。僵人综合征目前仍是一种临床诊断,具备3 项临床特征,即肌强直、痛性痉挛的临床表现,激动肌与拮抗肌持续不自主放电的典型肌电图表现,以及阳性的自身免疫相关抗体的血清学表现。与僵人综合征相关的自身免疫性相关抗体有GAD65、甘氨酸受体α1(GlyRα1)、两性蛋白(amphiphysin)、调节亚单位二肽基肽酶样蛋白6(DPPX6)和Gephyrin 抗体[38]。经典型僵人综合征最常见的抗体是GAD65 抗体,此类患者可同时合并肾细胞癌、胸腺瘤、淋巴瘤等,免疫调节治疗联合苯二氮类药物可改善症状[39]。伴强直和肌阵挛的进展性脑脊髓炎以肌强直、痛性肌痉挛、脑干脊髓症状、自主神经功能障碍、刺激诱发肌阵挛为临床特点,发病与GlyRα1 抗体密切相关,但该抗体亦可见于10%~15%的经典型僵人综合征病例,若实验室检测显示外周血GlyRα1 抗体阳性则提示免疫调节治疗疗效良好[40]。DPPX6 抗体阳性患者亦可以表现为伴强直和肌阵挛的进展性脑脊髓炎,持续积极的免疫调节治疗可以改善症状与体征[41]。两性蛋白抗体是诱发副肿瘤性僵人综合征、僵肢综合征和伴强直和肌阵挛的进展性脑脊髓炎的相关抗体,患者最常罹患乳腺癌[42],其次为非小细胞肺癌[43];此外,与两性蛋白抗体相关的神经系统疾病还有周围神经病、脑病、脊髓病和小脑共济失调。与经典型相比,副肿瘤性僵人综合征患者颈部和上肢僵硬更加显著,糖皮质激素治疗有效,且治疗原发肿瘤后症状可明显改善[42]。此外,Butler等[44]在1 例同时罹患僵人综合征和纵隔肿瘤患者的外周血中检测到Gephyrin 抗体,故认为该抗体也是介导副肿瘤性僵人综合征的相关抗体之一。
5.免疫相关神经性肌强直 免疫相关神经性肌强直(NMT)系周围神经过度兴奋引起的一种自发性持续肌肉收缩,典型表现为肌颤搐、肌痉挛、假性肌强直,常伴有出汗增多;当合并自主神经功能紊乱、中枢神经系统功能障碍如睡眠障碍和精神异常时,则称为Morvan 综合征。免疫相关神经性肌强直是临床最为常见的获得性神经性肌强直,亦称Issacs 综合征。Morvan 和Issacs 综合征均与电压门控性钾离子通道(VGKC)复合体抗体具有较高的相关性,尤其是LGI1 和CASPR2 抗体[45]。LGI1 抗体可见于Issacs 综合征患者,更常见于边缘性脑炎病例,患者在病程中可伴发面-臂肌张力障碍发作、低钠血症、近记忆力减退、睡眠障碍等症状,较少合并肿瘤,预后良好[6];CASPR2 抗体阳性主要见于Morvan综合征或Issacs 综合征,表现为边缘性脑炎、帕金森综合征、慢性神经病理性疼痛等症状,大多合并有胸 腺 瘤[6,46]。Irani 等[47]发 现,逾1/3 的Morvan 综 合征患者合并胸腺瘤,其中一些患者可同时检出AChR 抗体。
6.眼阵挛-肌阵挛综合征 眼阵挛-肌阵挛综合征(OMS)的特征性临床症状为眼阵挛,即双眼不自主、无规律、大幅度共轭快速眼动,尤以注意或追视物体时最显著,当注视目标固定时眼阵挛减轻,伴发症状以共济失调常见。研究显示,儿童眼阵挛-肌阵挛综合征可能与神经母细胞瘤相关[48];成人副肿瘤性眼肌阵挛-肌阵挛综合征(POMS)与多种自身免疫 性 相 关 抗 体 相 关,诸 如Ri、Hu、Yo、Ma1、Ma2、NMDAR、CRMP5 抗体等,可合并非小细胞肺癌、乳腺癌、卵巢癌等[49]。2016 年,Armangué等[50]共报告3 例眼阵挛-肌阵挛综合征合并肺癌病例,并且发现1 种新型的抗原决定簇——人类自然杀伤分子1(HNK-1)。眼阵挛-肌阵挛综合征相关病原体包括HIV 病毒、肺炎支原体、巨细胞病毒、人类疱疹病毒6 型、EB 病毒、西尼罗河病毒、丙型肝炎病毒等[49],单独应用糖皮质激素或免疫球蛋白或与抗菌药物联合应用均有效,表明眼阵挛-肌阵挛综合征与感染激活的免疫系统炎症反应有关。
7.其他类型免疫相关运动障碍 NF155 抗体被认为是慢性炎性脱髓鞘性多发性神经根神经病(CIDP)的致病性自身免疫性相关抗体,NF155 抗体阳性的慢性炎性脱髓鞘性多发性神经根神经病患者可发生感觉性共济失调和震颤,部分患者病程中可合并出现中枢和周围神经系统联合脱髓鞘疾病(CCPD)[51]。研究显示,约67% DPPX6 抗体阳性脑炎患者表现为中枢神经系统过度兴奋症状与体征,尤以运动障碍症状最为突出,如肌阵挛、多动和震颤,其他表现还有腹泻和(或)体重减轻、认知功能障碍、精神行为异常、癫发作等;患者对免疫治疗反应良好[52]。IgLON5 抗体相关脑病的重要特征为运动障碍,包括步态不稳、共济失调、构音障碍、舞蹈样动作、口-面部不自主运动等,亦可出现睡眠障碍如严重失眠、非快速眼动睡眠期行为障碍、睡眠呼吸暂停等,多数患者对免疫调节治疗效果欠佳[5]。
临床常见运动障碍自身免疫性相关抗体及临床特点参见表1。
二、免疫调节治疗性相关运动障碍
1.免疫调节治疗相关运动障碍 CAR-T 疗法是一项新型精准靶向治疗肿瘤的技术,神经毒性反应是其特有的药物不良反应,虽然仅发生在部分细胞因子释放综合征(CRS)患者,但亦有极少数患者在无细胞因子释放综合征的情况下并发神经系统不良事件[53]。约有32%~64%的肿瘤患者于CAR-T 治疗后5 ~7 天发生神经毒性反应,表现为震颤、运动障碍等,严重者可进展为局灶性神经功能缺损或全面性强直-阵挛发作(GTCS)等脑病[54-55]。PD1 是重要的免疫抑制因子,与组织细胞表面的细胞程序性死亡蛋白配体1(PDL1)相结合,抑制淋巴细胞功能,因此,PD1 抑制剂可通过阻断与肿瘤细胞的PDL1 结合增强对肿瘤细胞的免疫反应。与PD1 抑制剂有关的神经系统并发症发生率约4.2%[56],以神经-肌肉并发症较为常见,亦有PD1 抑制剂导致运动障碍、小脑共济失调的文献报道[57]。
2.免疫重建炎性综合征相关运动障碍 HIV 感染患者经高效抗逆转录病毒疗法治疗后,在其免疫功能重建过程中出现的临床表现恶化现象,称为免疫重建炎性综合征(IRIS),发病机制尚未阐明,可能与机体免疫功能失调等多种因素有关。免疫重建炎性综合征相关运动障碍主要表现为眼肌阵挛-肌阵挛综合征,通常发生于CD4+T 细胞数目迅速增加的背景下或经高效抗逆转录病毒疗法治疗后,提示眼肌阵挛-肌阵挛综合征可能是免疫重建炎性综合征的一种特殊表现[58]。
三、小结
神经系统自身免疫性疾病的临床表现多样,特别是运动障碍的表现形式更加复杂,其病理生理学机制与传统神经变性病导致的运动障碍不尽相同,治疗方案更是多样。临床遇到呈急性或亚急性发病的运动障碍,更应考虑自身免疫性疾病所致。大部分自身免疫性疾病导致的运动障碍在消除病因后症状可以部分甚至完全改善,即所谓的“可逆性”。治疗原则采取以免疫调节治疗为主的综合治疗,有别于神经变性病所致运动障碍的“不可逆性”和“对症治疗”。
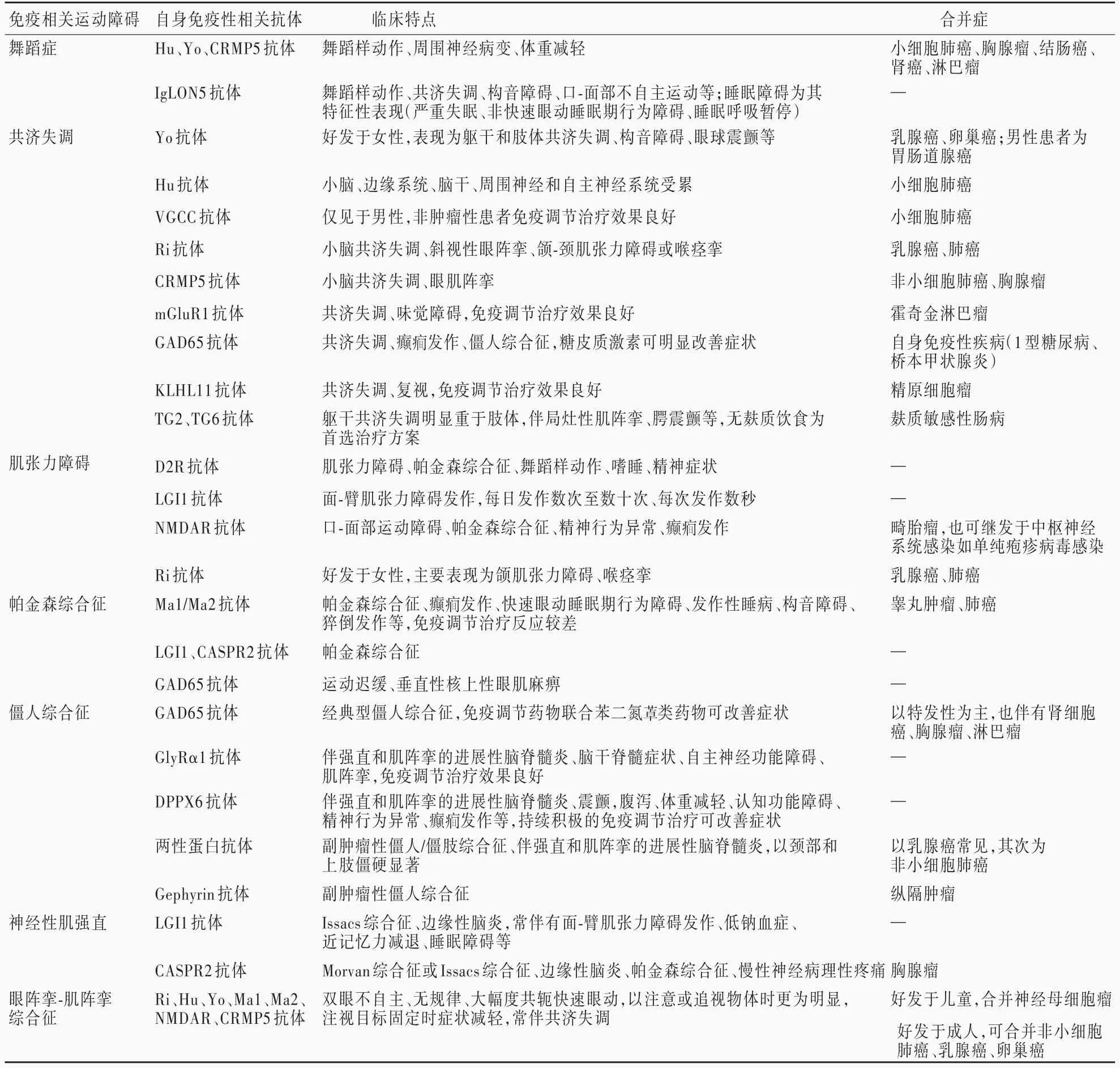
表1 运动障碍自身免疫性相关抗体及临床特点Table 1. Autoimmune antibodies and clinical features of movement disorder
由于运动障碍是神经系统自身免疫性疾病重要的甚至是首发的临床表现,因此提高对此类运动障碍的诊断与鉴别诊断能力有助于提升临床医师对神经系统自身免疫性疾病的认识。神经系统自身免疫性疾病相关运动障碍有可能将成为运动障碍家族中最特殊的“分支”,随着相关研究的开展,其神秘面纱将逐渐被临床医师揭开。
猜你喜欢
中国神经精神疾病杂志(2022年3期)2022-07-14
广东海洋大学学报(2022年2期)2022-03-31
浙江临床医学(2022年2期)2022-03-19
保健与生活(2021年15期)2021-08-16
湖南农业大学学报(自然科学版)(2021年4期)2021-08-14
放射学实践(2021年5期)2021-05-21
医学概论(2021年19期)2021-01-21
世界科学技术-中医药现代化(2020年2期)2020-07-25
祝您健康·文摘版(2020年5期)2020-05-13
中西医结合心血管病电子杂志(2016年9期)2016-11-16
