
AvrLr19 突变菌株诱导TcLr19 小麦中 差异表达基因分析
2020-10-30 03:01崔钟池武文月王婉晴胡铁猛王海燕刘大群
河北农业大学学报 2020年5期
崔钟池,武文月,王婉晴,胡铁猛,王海燕,刘大群,2
(1.河北农业大学 植物保护学院/河北省农作物病虫害生物防治技术创新中心,河北 保定 071000; 2.中国农业科学院 研究生院,北京100081)
小麦叶锈菌(Puccinia triticina, Pt)引起的小麦叶锈病是世界范围内发生最普遍,对经济影响最严重的小麦病害之一,引入抗病基因是应对小麦叶锈病的根本所在。然而,单一的抗病品种导致寄主选择压力,叶锈菌毒性变异频繁,新的毒性小种不断出现,往往导致小麦抗叶锈病品种抗性丧失。于是,深入探究小麦抗病相关基因,并明确它们的功能及其参与的抗病信号通路,对于发掘新的抗病基因及揭示寄主—病原物的互作机制具有重要的理论和 实践意义。
转录组测序技术RNA-seq 已成为基因表达分析的首选工具[1],该技术被广泛应用于鉴定小麦对生物胁迫的响应基因及其代谢通路,通过分析寄主—病原物间亲和与非亲和互作反应中的差异表达基因,评估病原物是如何调节致病基因的表达以及如何影响寄主植物的防御反应[2],为发掘抗病相关基因或致病因子提供了更加快捷的途径。张艳俊等[3]对接种叶锈菌PHNT 后的小麦材料‘TcLr19’进行转录组测序,筛选到了4 个与抗病相关的候选基因(NAC 类转录因子、bZIP 型转录因子、丝氨酸/苏氨酸激酶和酪氨酸蛋白激酶);武文月等[4]基于PHNT 接种‘TcLr19’转录组数据筛选到24 个叶锈菌PHNT 候选效应蛋白;Wu 等[5]对携带Lr47 的小麦品种“Lr47-Yecora Rojo-BC6F5”、“Lr47-UC1037”和“Lr47-White Yecora”进行RNA-seq分析,发现了863 个显著上调基因和418 个显著下调基因,发现Ca2+通道基因(CNGCs、CDPK、Rboh、CaM)、FLS2、RPM1、PR1 及HSP90 等 多个抗病基因富集显著。此外,区别于以上分析病原菌侵染遗传背景不同的感、抗寄主品种的转录组数据,甲基磺酸乙酯(EMS)化学诱变与转录组结合是研究真菌与寄主抗病相关基因关系的有效方法,被广泛用于克隆植物宿主的抗性基因[6]。基因突变被证明是引起病原菌变异的主要机制[7],EMS 是研究各种生物体突变时最常用的诱变剂,可引起碱基C/G 到T/A 的变化[8]。利用EMS 人为使病原菌发生突变后,分析病原物突变前后对同一抗病品种中基因表达的不同影响,为抗病基因的筛选甚至小种毒性变异机制提供更加精确的研究方向。Salcedo 等[9]和Chen 等[10]利用EMS 获得小麦秆锈菌突变体,分别从小麦秆锈病病原中获得了无毒基因AvrSr35 和AvrSr50,极大地促进了锈菌无毒基因的研究进展。
小麦抗叶锈病基因Lr19 来源于长穗偃麦草(Agropyron elongatum),定位于7D 染色体上[11], 在小麦全生育期表现高抗性。栗小英[12]、高琳等[13]先后从TcLr19 中克隆出抗病相关基因 TaTLP1 与TaPR1;张家瑞等[14]利用病毒介导基因 沉 默(Virus-induced Gene Silencing , VIGS)技术证明TaTLP1 具有抗叶锈病功能;梁芳等[15]利用农杆菌介导法鉴定了TaPR1 的抗病功能,王菲等[16]利用酵母双杂交(Yeast two-Hybrid Assay,Y2H)、 双 分 子 荧 光 互 补(Bimolecular fluorescent complimentary, BiFC)和免疫共沉淀(Co-immunoprecipitation, Co-IP)明确了TaTLP1与TaPR1 相互作用。以上研究表明Lr19 在小麦抗叶锈病研究方面有巨大应用潜力。本课题组前期利用EMS 诱变获得使小麦TcLr19 感病的叶锈菌PHNT 毒性突变菌株[17],并证实发生突变的位点为AvrLr19。在此基础上,本研究将野生型PHNT与PHNT 突变体分别接种TcLr19 进行转录组测序,以期获得小麦TcLr19 中在不同反应型下的差异表达基因,通过对差异表达基因的GO(Gene Ontology)及KEGG(Kyoto Encyclopedia of Genes and Genomes)富集分析,初步明确TcLr19 中抗病相关基因的种类与功能,以及抗叶锈菌相关的代谢通路,为进一步研究抗叶锈基因Lr19 的抗病机制奠定基础。
1 材料与方法
1.1 植物材料与菌株
植物材料为小麦抗叶锈病近等基因系材料‘TcLr19’;菌株为野生型小麦叶锈菌07-10-426-1(PHNT),以及EMS 处理后的PHNT 突变体(命名为‘M-PHNT’),均由河北农业大学植物病害生物防治和分子植物病理学实验室提供。
1.2 方法
1.2.1 样本处理 采用扫苗法分别接种PHNT 及M-PHNT的夏孢子于一叶一心期的小麦材料‘TcLr19’上,剪取接菌14 d 后的叶片,每组样本设3 个生物学重复。PHNT 接种TcLr19 作为非亲和互作样本组,命 名 为“WT(WT_sample1,WT_sample2,WT_sample3)”,M-PHNT 接种TcLr19 作为亲和互作样本组,命名为“MT(MT_sample4,MT_sample5, MT_sample6)”,用液氮将其迅速冷冻,储藏于-80 ℃ 冰箱中,备用。
1.2.2 RNA-seq 及原始数据处理 将准备好的样品送上海欧易生物医学科技有限公司进行文库构建和测序工作。利用hisat2[18]将Clean reads 分别与中国春小麦基因组序列草图(the bread wheat cultivar Chinese spring)和小麦叶锈菌BBBD 数据库进行序列比对,获取在参考基因组或基因上的位置信息,利用R 语言进行Pearson 相关系数计算,检测样品间相关性。
1.2.3 差异表达基因分析 利用DESeq 软件对各个样本基因的counts 数目进行标准化处理(采用basemean 值来估算表达量),计算差异倍数,并采用NB(负二项分布检验的方式)对reads 数进行差异显著性检验,最终根据差异倍数及差异显著性检验结果来筛选差异蛋白编码基因,筛选差异的条件为差异显著性(P-value)<0.05 且差异倍数(Foldchange)>2。通过R 语言中的ggplot2绘制火山图和MA 图了解差异表达基因的整体分布情况。
1.2.4 差异表达基因GO 和KEGG 分析 对差异表达基因进行GO 富集分析[19],结合GO 数据库,http://www.geneontology.org/)对其功能进行描述。利用KEGG 数据库(http://www.genome.jp/kegg/)对差异蛋白编码基因进行代谢通路(Pathway)分 析[20],并用超几何分布检验的方法计算每个Pathway 条目中差异基因富集的显著性。计算的结果会返回一个富集显著性的P-value,较小的P-value表示差异基因在该Pathway 中出现显著富集。
2 结果与分析
2.1 测序reads 基因组比对结果及样本相关性分析
将reads 与指定的参考基因组进行序列比对, 获得WT 和MT 样本比对上寄主小麦及叶锈菌参考序列比率信息(图1A)。WT 样本组,寄主比对上参考基因组的序列所占百分比分别为89.34%、87.49%和90.16%,叶锈菌比对上参考基因组的序列所占百分比分别为5.33%、4.18%和2.53%;MT样本组,寄主比对上参考基因组的序列所占百分比分别为65.04%、48.74% 和58.37%,叶锈菌比对上参考基因组的序列所占百分比分别为20.16%、32.3% 和26.87%。分析结果显示,野生型叶锈菌株突变后对Lr19 产生毒性,成功侵染抗病品种‘TcLr19’。根据基因表达情况获得测序样品间的相关系数图如图1B 所示,对于整体基因表达水平,在生物学重复中观察到明显的相关性。以上结果表明,送测样本合理,转录组测序结果可靠,可进行深入的分析。

图1 测序reads 基因组比对结果及样本相关性分析Fig. 1 Genome comparison results and sample correlation analysis of sequencing reads
2.2 差异表达基因筛选
转录组测序结果显示,在WT 中共检测到 66 768 个基因表达,在MT 中检测到68 383 个基因表达,以P-value <0.05 且Foldchange >2 作为筛选标准,2 个样本组中共筛选到2 114 个差异表达基因(图2A),其中有1 189 个上调表达基因,925个下调表达基因(图2B)。MA 图(图2C)和火山图(图2D)反映了差异表达基因整体分布情况。结果表明,非亲和反应中有大量抗病相关基因上调表达,笔者将重点分析差异表达基因中的上调表达基因。
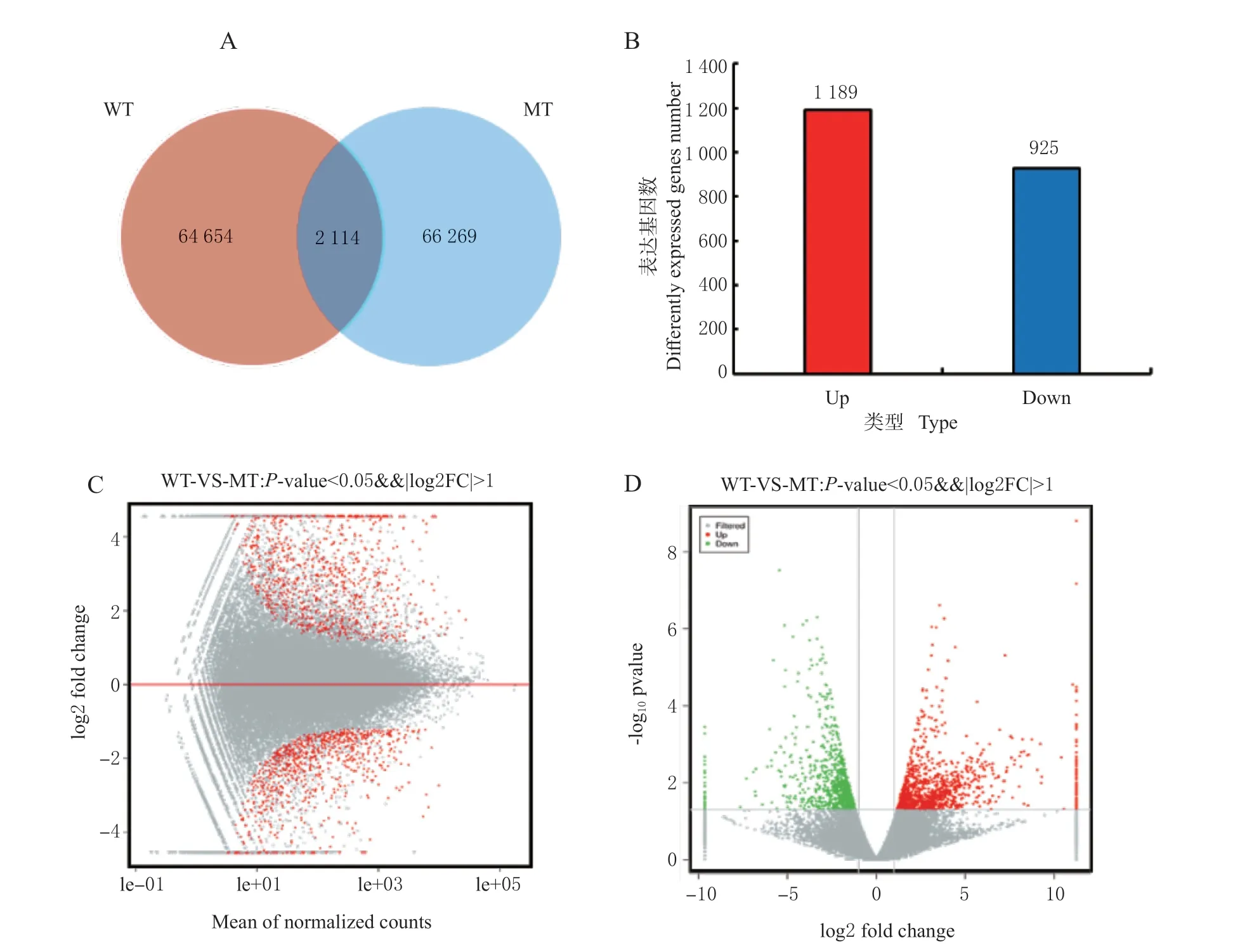
图2 WT-VS-MT 差异表达基因统计(差异倍数大于2,差异显著性小于0.05)Fig. 2 DEGs in WT-VS-MT (fold change > 2, P-value < 0.05)
2.3 差异表达基因GO 和KEGG 分析
对1 189 个上调差异表达基因进行GO 富集分析,上调的差异表达基因被注释为氧化还原、真菌防御、过氧化氢酶活性和几丁质酶活性等功能。对GO 注释条目进行进一步筛选,列举出部分GO 分类条目中的抗病相关基因(见表1),在WT 样本小麦中β-1,3-葡聚糖酶以及过氧化氢酶等基因上调表达,几丁质酶表达数量较多,这些结果表明了以上基因和小麦抗病反应有密不可分的关系。
其中过氧化氢酶、几丁质酶和脂肪酰基辅酶A 还原酶分别被富集到过氧化物酶(Peroxisome,ko04146)、氨基糖/ 核苷酸糖代谢(Amino sugar and nucleotide sugar metabolism,ko00520)和角质生物合成(Cutin biosynthesis,ko00073)3 个代谢通路中(表1),说明这些信号通路参与了TcLr19抗叶锈病过程。
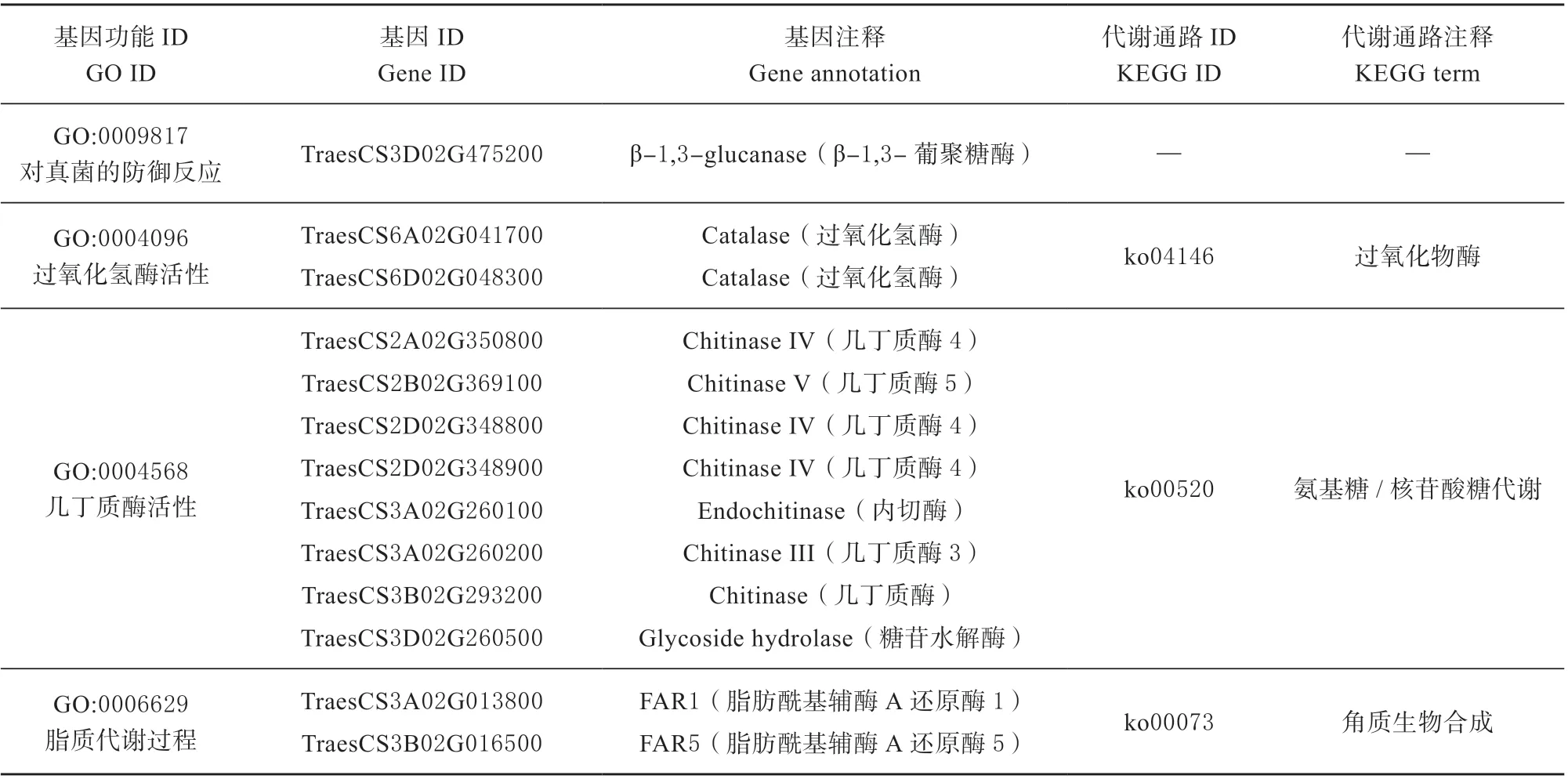
表1 WT-VS-MT 差异表达基因GO 和KEGG 分析Table 1 GO and KEGG analysis of WT-VS-MT DEGs
2.4 叶锈菌诱导位于7D 染色体上的差异表达基因
由于携带Lr19 基因的染色体被证实是普通小麦中的7D 染色体,因此本研究重点对转录组数据中位于7D 染色体上的差异表达基因进行了探索。与亲和互作组相比,许多位于7D 染色体上的基因被诱导差异表达,共鉴定了10 个差异表达基因(表2),利用Mapchart 软件绘制了其在7D 染色体上的定位(图3)。醛脱氢酶2C4、细胞色素P450、病程相关蛋白1 ~4、乙烯转录因子和热激蛋白基因在WT 中的表达量高于MT 组;另外,本研究发现,有5 个基因TraesCS7D02G027600、TraesCS7D02G320200、TraesCS7D02G353900、TraesCS7D02G387000 和TraesCS7D02G479300,分别编码Fe2+吸收转运基因(MEBL)、蛋白连接酶(FBL23)、醛脱氢酶,半胱氨酸活性位点(ALDH2C4)、二酰基甘油激酶(DGK5)和GTP 结合蛋白(OBGM),这5 个基因只在非亲和互作组中表达。以上结果表明,7D 染色体上的许多基因在小麦抗病反应中扮演了重要的角色,尤其是在非亲和反应中特异表达的5 个基因发挥了更重要的抗病功能。
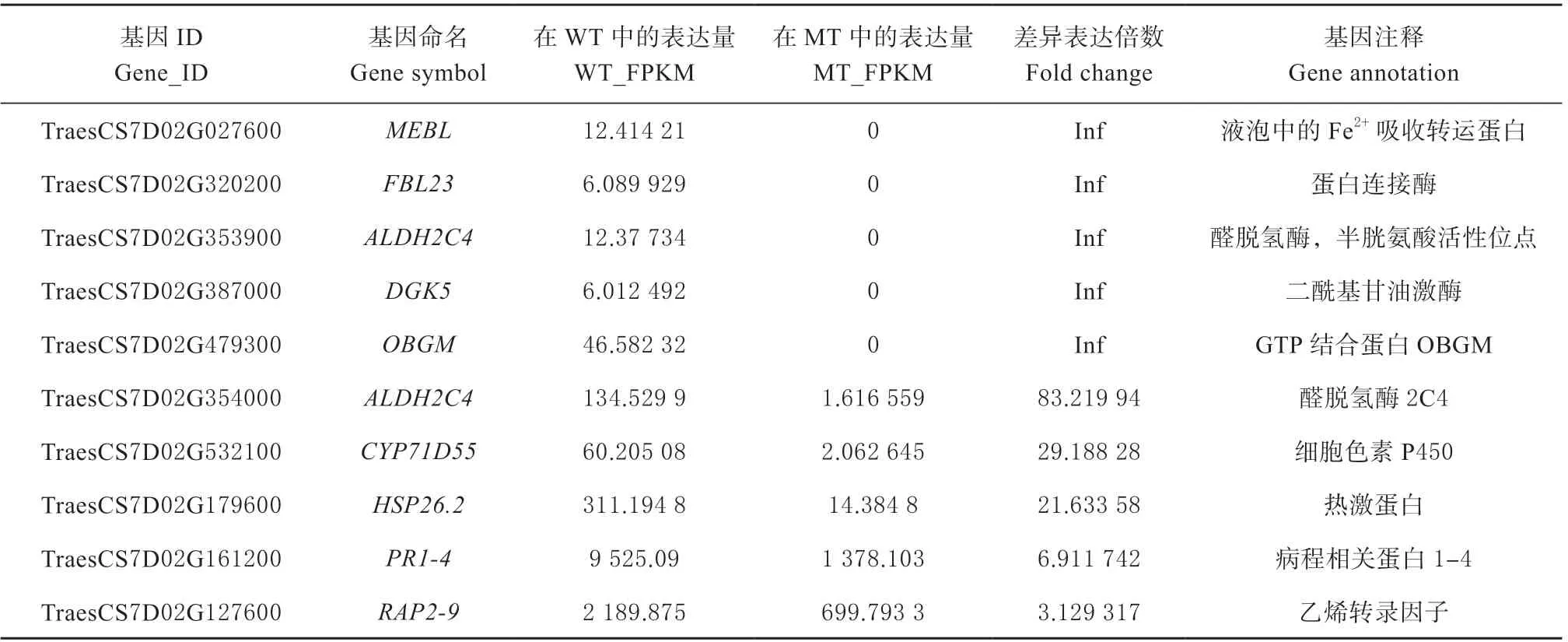
表2 定位于7D 染色体上的差异表达基因(WT-VS-MT)Table 2 DEGs located on chromosome 7D( WT-VS-MT)

图3 WT-VS-MT 差异表达基因7D 染色体定位Fig.3 Distribution map of DEGs located on chromosome 7D
3 讨论与结论
植物受到生物胁迫时,植物体内的防御反应基因的表达模式就会发生变化,起到抵抗外界物质侵染的作用。根据“基因对基因(gene-for-gene)”学说[21],病原物无毒基因和毒性基因会对植物体内基因的表达情况产生不同的影响,在非亲和互作反应中,植物体内的抗病相关基因会上调表达。张艳俊[22]利用RNA-Seq 分析了叶锈菌PHNT 接种TcLr19 后不同时间点的基因表达情况,发现了几丁质酶活性和转录因子活性基因在特定的时间内上调表达。范学峰[23]用叶锈菌THTT 分别接种TcLr19 和TcLr19 突 变 体,RNA-Seq 分 析 结 果 显示在TcLr19 抗病反应中氧化还原代谢通路富集显著。与以上研究不同的是,本研究针对田间尚未发现Lr19 毒性叶锈菌菌株的现状,在利用EMS 创制了对TcLr19 具有毒性的PHNT 突变体的基础上,进行全基因组范围抗病相关基因的筛选。PHNT 突变体仅针对Lr19 毒性位点发生了改变,对比PHNT 和M-PHNT 两种菌株分别诱导TcLr19 中的差异表达基因,增强了对Lr19 抗病及抗病相关基因研究的目的性。当然,植物体内除被病原物诱导表达的基因外,还存在一部分组成型表达的抗病相关基因,比如小麦中S2A2 基因[24]。所以,想要全面筛选Lr19 抗病及抗病相关基因,只分析差异表达基因是不够的,在后续研究中,会同时对两组中的非差异表达基因进行筛选分析,完善对Lr19 抗病机制的研究。
GO 分析结果显示,部分差异表达基因被注释为β-1,3-葡聚糖酶、几丁质酶等功能。几丁质和β-1,3-葡聚糖是真菌细胞壁的主要组成物质,几丁质酶和β-1,3-葡聚糖酶协同表达时,可以分解病原真菌的细胞壁以抵御侵害[25],本课题组在前期研究中也报道β-1,3-葡聚糖酶基因的表达明显受叶锈菌的诱导,并参与了小麦抗叶锈病防御反应[26]。本研究中发现了新的抗病相关基因被诱导上调表达,GO富集分析为TcLr19 中抗病相关基因的筛选缩小了范围,在下一步工作中将进行表达分析和功能验证。
KEGG 可以将基因与基因表达信息作为一个整体网络进行研究,为明确抗病相关代谢通路和系统筛选抗病的相关基因提供思路。余颖豪等[27]通过对金针菇菌丝阶段和原基形成阶段的转录组分析,明确了金针菇冷诱导形成原基的分子机制及调控 网络。在KEGG 富集结果中,发现了过氧化物酶、氨基糖/ 核苷酸糖代谢和角质生物合成3 个代谢 通路在WT 非亲和互作反应中富集显著,与张绳 敏[28]、柴乖强[29]等先前研究报道的过氧化物以及角质生物合成相关基因参与小麦抗病反应结果相符。KEGG 分析初步明确了以上3 个代谢通路参与了TcLr19 的抗病过程,接下来会继续关注与这些代谢通路相关的基因和通路,探索TcLr19 的抗病代谢网络。
此外,本研究对与Lr19 同在7D 染色体上的差异表达基因进行专项分析,筛选出7D 染色体上的部分抗病相关基因,其中有5 个基因在抗病反应中特异性表达,有可能抗病关系更加密切,后续将对其进行生物信息学、qRT-PCR 以及功能验证,以确定其在TcLr19 抗叶锈病防御反应中的作用,有助于进一步分析Lr19 对抗病相关基因的调控。
猜你喜欢
作物学报(2022年11期)2022-08-31
今日农业(2022年4期)2022-06-01
西北农业学报(2022年4期)2022-05-19
西北农业学报(2021年12期)2022-01-20
农业技术与装备(2021年12期)2021-12-02
湖北农业科学(2021年10期)2021-06-25
世界农药(2020年4期)2020-12-22
农业科技与信息(2020年20期)2020-12-18
发明与创新·大科技(2019年5期)2019-07-31
新农村(2018年35期)2018-04-02
