
Interaction of 1, 3, 3-Trinitroazetidine (TNAZ) and Zinc
——A DFT Treatise
2020-10-28 01:25Lemirker
火炸药学报 2020年5期
Lemi Türker
(Middle East Technical University, Department of Chemistry üniversiteler, Eskisehir Yolu No.1, 06800 Çankaya/Ankara, Turkey)
Abstract:In order to investigate whether zinc atom is compatible with 1, 3, 3-trinitroazetidine (TNAZ) structure, TNAZ+Zn and 2TNAZ+Zn composite systems have been considered within the constraints of density functional theory at the level of B3LYP/6-31++G(d,p) and ωB97X-D/6-31++G(D,P). In the case of TNAZ+Zn composite the both level of calculations resulted that one of the C—NO2 bonds of TNAZ undergoes bond cleavage. However, as the zinc content decreases by increasing the TNAZ content, C—NO2 bond elongation considerably decreases, although it is still longer than the similar bonds in 2TNAZ+Zn composite in which zinc, by weight, is 7.14%. In all the cases the zinc atom acquires positive charge. The results indicate that the interfrontier molecular orbital energy gaps (HOMO-LUMO energy difference) of the composites decrease as the zinc content decreases.
Keywords:quantum chemistry;1, 3, 3-trinitroazetidine;TNAZ;explosive;zinc;DFT
Introduction
An energetic small-ring compound 1, 3, 3-trinitroazetidine, also known as TNAZ, is the most widely studied explosive recently because of powerful but insensitive explosives are continuously searched theoretically and experimentally[1-2]. Its performance as an explosive is said to be in between hexogen (RDX) and octogen (HMX) but it is considerably less sensitive[3]. It possesses a highly nitrated four-membered nitrogen heterocyclic ring (azetidine ring) having both C—NO2and N—NO2groups. Its improved performance in comparison to conventional trinitrotoluene (TNT), a melt castable explosive, is attributed to the presence of strained ring system (azetidine ring)[4-9]. Various methods have been reported for the synthesis of 1, 3, 3-trinitroazetidine[10].
Note that TNAZ, being a high performance, melt castable explosive, has been proposed as potential replacement for TNT[11]. The low melting point of TNAZ (101℃) allows the processing of various formulations on modified production lines. It has approximately 30% greater performance than TNT. It also shows excellent thermal stability (>180℃)[12]. TNAZ has many additional advantages over the known explosives. It is a highly energetic material, more powerful than RDX and is less vulnerable than most other nitramines[13-14]. Unlike HMX, TNAZ is soluble in molten TNT, and additionally it is compatible with aluminum, steel, brass and glass[15-17].
By using the pressure DSC method the compatibility of 1,3,3-trinitroazetidine (TNAZ) with some energetic components and inert materials of solid propellants has been studied[18]. On the other hand, desensitization of TNAZ via molecular structure modification has been recently investigated theoretically[19].
Reactive molecular dynamics simulations of the thermal decomposition mechanism of TNAZ has been studied where the thermal decomposition of its crystals at high temperature was calculated by molecular dynamics simulation with the ReaxFF/lg reactive force field[20]. Also the change in the potential energy of TNAZ, the formation of small-molecule products and clusters, and the initial reaction path of TNAZ were analyzed. Moreover, the kinetic parameters of different reaction stages in thermal decomposition of TNAZ were obtained[20].
Aluminum and some other metal powders are added to explosives in order to enhance their blast and heat effects, as well as to increase the bubble energies in underwater explosions[21-25]. Therefore, aluminized explosives have been used in various formulations since the beginning of the last century. However, zinc is not employed often as explosive component not only because of lesser heat of formation value of its oxide but may be due to some incompatibility reasons. Some computational studies have revealed that Cl-20 and TEX are to be affected by zinc[26-27].
In the present study,effect of zinc on TNAZ has been investigated at the molecular level within the constraints of density functional theory (DFT).
1 Method of calculation
In the present study, the initial geometry optimizations of all the structures leading to energy minima have been achieved by using MM2 method followed by semi-empirical PM3 self-consistent fields molecular orbital (SCF MO) method[28-29]at the restricted level[30-31]. Subsequent optimizations were achieved at Hartree-Fock level using various basis sets. Then, geometry optimizations were managed within the framework of density functional theory (DFT)[32-33]using two different functionals and the same basis set B3LYP/6-31++G(D,P) andωB97X-D/6-31++G(D,P)[31,34-36]. The exchange term of B3LYP consists of hybrid Hartree-Fock and local spin density (LSD) exchange functions with Becke′s gradient correlation to LSD exchange[35,37]. The correlation term of B3LYP consists of the Vosko, Wilk, Nusair (VWN3) local correlation functional[38]and Lee, Yang, Parr (LYP) correlation correction functional[39]. Also, the vibrational analyses were done. The total electronic energies are corrected for the zero point vibrational energy (ZPE). The normal mode analysis for each structure yielded no imaginary frequencies for the 3N—6 vibrational degrees of freedom, where N is the number of atoms in the system. This indicates that the structure of each molecule corresponds to at least a local minimum on the potential energy surface. All these calculations were done by using the Spartan 06 package program[40].
2 Results and discussion
In order to investigate the compatibility of zinc atom with 1, 3, 3-trinitroazetidine (TNAZ) structure, TNAZ+Zn and 2TNAZ+Zn composite systems have been investigated. Two different functionals with the same basis set have been used to visualize whether any parallelism exists or not on the effect of zinc. Functional,ωB97X-D is a new class of DFT functional known as long-range separated functional which is capable of capturing both short-range and long-range interactions. On the other hand, B3LYP can only capture short-range interactions but it is generally much faster than most post-Hartree-Fock techniques and usually yields comparative results[41].
Zinc atom has ground state electronic configuration of 1s22s22p63s23p63d104s2. It participates various inorganic and organic reactions, especially as reducing agent or forming some organozinc compounds[42]. Although, aluminum is incorporated in some ammunition formulations or explosive compositions, zinc is not employed (up to the best knowledge of the author). The idea of using some metals in those formulations is to increase the thermal devastating effect which is due to heat evolved as certain metal oxide formations occur. In that sense, aluminum should be more effective than zinc. The heat of formation values of Al2O3and ZnO are-1669kJ/mol and -348kJ/mol[43], respectively. The other reason for zinc not to be preferred in some ammunition formulations or explosive compositions could be its stronger reducing power. It has strong power of reducing nitro compounds[42].
On the other hand, TNAZ molecule having three nitro groups in its structure such that one of them is a nitramine type is a potent molecule to be attacked by zinc. Theoretically it would be interesting to find out which type of bond is susceptible to the effect of zinc.
In the present study, TNAZ+Zn composite system (C1, C2 conformers) and observing its instability, 2TNAZ+Zn composite system (D) have been subjected to computational treatment.
Figure 1 shows the optimized structures of these composites. Note thatωB97X-D/6-31++G(d,p) level of optimization of C2 failed to produce a different geometry than C1 conformer at that level. Therefore, any relevant part is missing in various
Figures in the present study. Also it is worth mentioning that in the case of D (2TNAZ+Zn) the optimized structures are quite dissimilar (in relative orientation of the components) at two different levels of calculations performed presently (see
Figure 1).
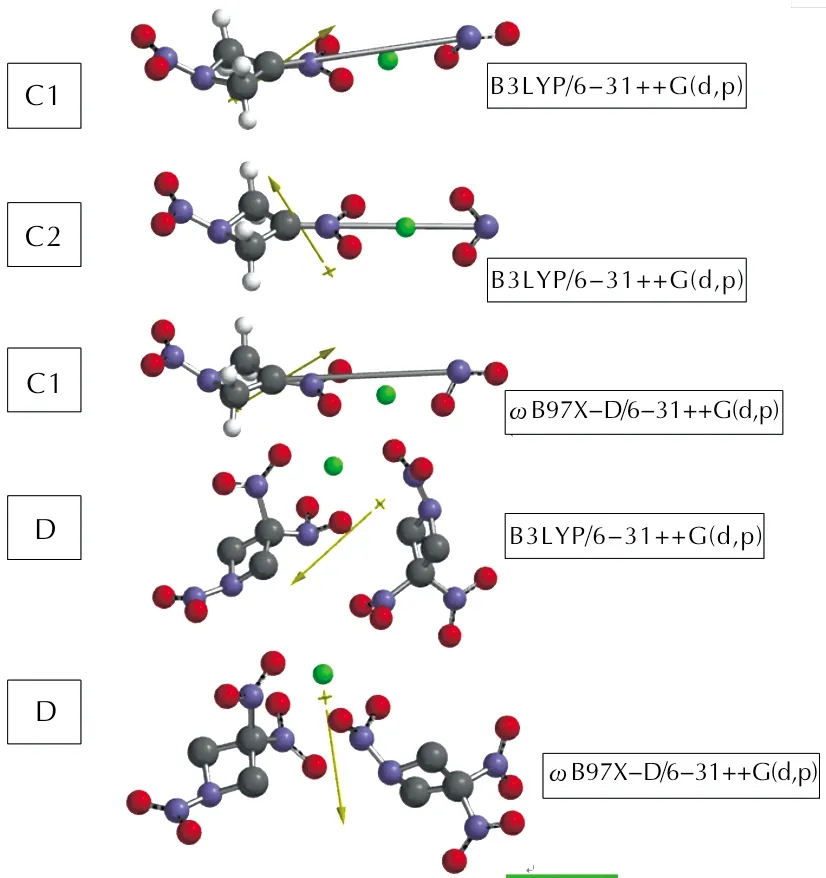
Fig.1 Optimized structures of the composites considered
The orientation of TNAZ components in D is noticeable. That C—NO2bonds of one component is next to the N—NO2bond of the other. Note that the Zn contents of the composites considered presently are 25.39 % (C1,C2) and 7.14 % (D) by weight, respectively.
In the case of TNAZ+Zn composite, two conformational forms have been detected at the level of B3LYP/6-31++G(d,p). and they are labeled as C1 and C2 of which C2 (as shown later in Table 1) is more stable conformer.
Figure 1 also shows the direction of the dipole moments of the composite systems. As seen in the
Figure, of the three nitro groups of TNAZ molecule, only one of the C—NO2bonds undergoes bond cleavage, the nitramine bond remains intact. Whereas, optimization of higher zinc contained composite, TNAZ+2Zn, failed.
In order to confirm that only one of the C—NO2bonds, but not the others, undergoes cleavage, alsoωB97X-D/6-31++G(d,p) type calculations have been performed (see
Figure 1) which yields a C1 like optimized structure. Note that in the case of B3LYP/6-31++G(d,p) level of calculations two conformers on the potential energy surface had been detected for TNAZ+Zn composite during the optimization, whereas due to the annihilation of the organic component the optimization process on TNAZ+2Zn composite failed. AlsoωB97X-D/6-31++G(d,p) level of calculations on TNAZ+Zn composite detected only C1 conformer.
Figure 2 shows the bond lengths of some of the composites. Although in C1 and C2 cases an NO2moiety is expelled from TNAZ, lower zinc percent having composite, 2TNAZ+ Zn system (D), is optimized successfully. However it has some elongated bonds. In one of TNAZ components of structure-D, one of C-NO2bonds is noticeable with a bond length of 1.64Å (Figure 2). Note that N-NO2bonds in D are in between 1.33Å—1.38Å and C—NO2bonds (except the particular C—NO2bond mentioned above) vary in between 1.41Å and 1.52Å. The shortest C—NO2bond is linked to the carbon atom also having the elongated C—NO2bond. Also note that the shortest and the longest C—NO2bonds belong to the same TNAZ component. Generally, in the literature C—NO2bond lengths are about 1.50—1.52Å depending on whether electron drawing or donating groups present on the same carbon, e.g.; 1.51Å (CH3)2C(NO2)2); 1.526Å (C(NO2)4); 1.592Å (CBr3NO2)[44]. The C—NO2bond of 1.64Å in composite D is some what longer as compared to above values. On the other hand, 3TNAZ+2Zn composite is also found to be unstable. Again one of C—NO2bonds of one of TNAZ components undergoes cleavage. Note that the zinc contents of the composites,nTNAZ:mZn, are 25.39 (1∶1); 18.42 (3∶2) and 7.14 (2∶1).
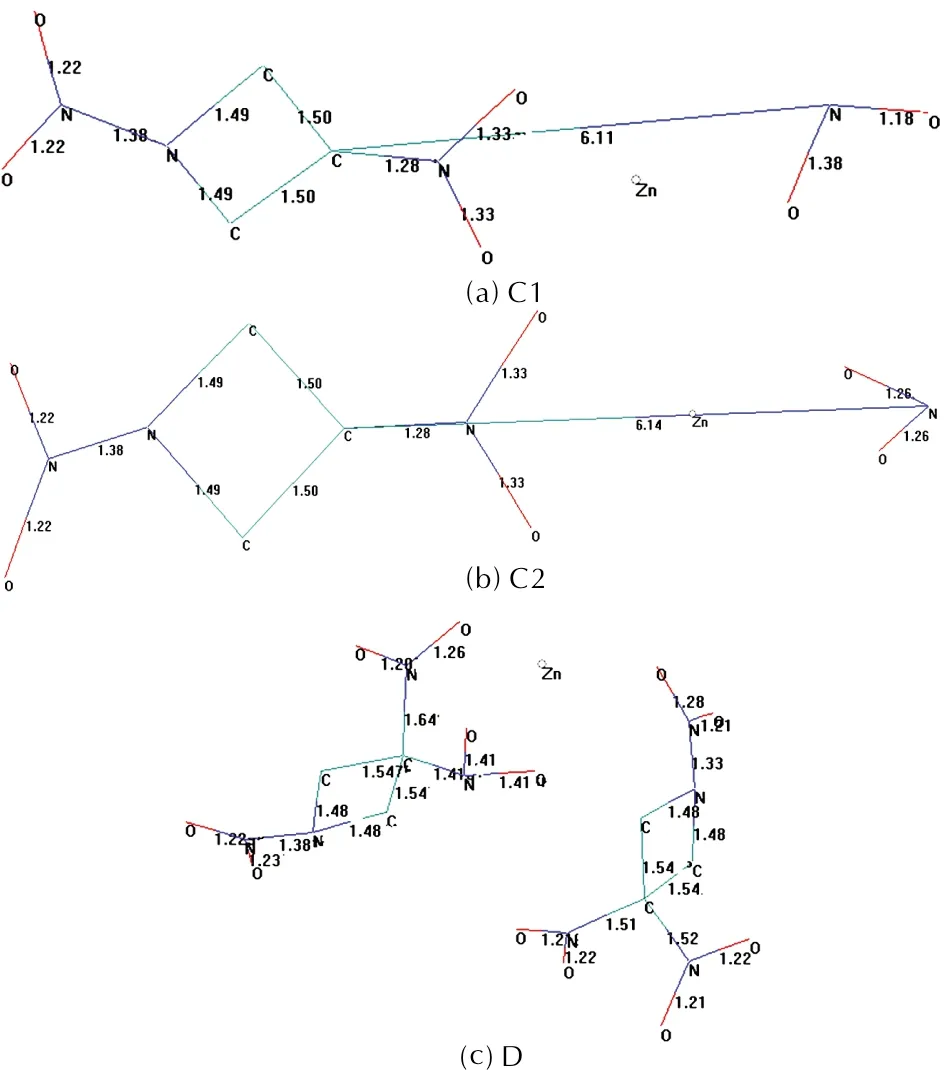
Fig. 2 Bond lengths (Å) of some of the composites considered (B3LYP/6-31++G(d,p))
Figure 3 shows electrostatic potential (ESP) charges on the atoms of some of the composites considered. Note that the ESP charges are obtained by the program based on a numerical method that generates charges that reproduce the electrostatic potential field from the entire wavefunction[40]. As seen in the
Figure the zinc atom acquires positive charge in each case. The NO2moiety departed from TNAZ component has over all negative charge in C1 and C2, however it is more negative in C1 case.

Fig. 3 Electrostatic potential charges of some of the composite structures (B3LYP/6-31++G(d,p))
Note that the positive charge of zinc atom in C1 and D are quite comparable. However, D seems to be comparatively more stable because it keeps its integrity. Most probably, the zinc atom donates some electron population to both of TNAZ components, of course in unequal extents. Thus the electron population gained by each component cannot cause as much elongation as it has been the case in C1 and C2.
Figure 4 shows the IR spectra of the composites presently considered. The peak of C1 at 1807cm-1is due to C—NO2stretching which is followed by peaks at 1697cm-1(N—O sym stretching of the departed NO2) and 1629cm-1(N—O asym stretching of N-NO2). In the case of C2 , the peak at 1870cm-1is C—NO2stretching followed by N—O sym stretching of the departed NO2at 1746cm-1and 1699cm-1N—O asym stretching of N—NO2bond. The peaks below 1500cm-1are certain bending vibrations and stretchings coupled with each other.
In the case of 2TNAZ+Zn composite, the peaks at 1671cm-1, 1621cm-1are asym N—O stretchings of C—NO2groups. The asym N—O stretchings of N—NO2groups occur at 1593cm-1.

Fig.4 IR spectra of TNAZ and the composites considered (B3LYP/6-31++G(d,p))
Table 1 shows some energies of TNAZ+Zn composites considered. As seen there conformer C2 is more stable than C1

Table 1 Some energies of the composite TNAZ+Zn
Figure 5 shows some of the molecular orbital energy levels of the composites of present concern. The more stable conformer, C2, is characterized with lower LUMO and HOMO energies as compared to those of conformer-C1.

Fig.5 Some of the molecular orbital energy levels of the composites (B3LYP/6-31++G(d,p))
Table 2 shows the HOMO, LUMO energies and the interfrontier molecular orbital energy gaps (Δε) of the composites considered as well as for TNAZ and 2TNAZ systems . The order of HOMO and LUMO energies are TNAZ<2TNAZ
Figure 2 and Table 2, as a conjecture, decrease of the zinc content helps to keep the integrity of TNAZ structure in the composites but they become more vulnerable to impact.

Table 2 The HOMO, LUMO energies and the molecular orbital energy gaps of the composites.
Figure 6 stands for the HOMO and LUMO patterns of the composites. In the case of C1 the HOMO is over the departing NO2moiety (at the both levels of calculations) whereas the LUMO spread over the remaining part of the decomposed composite. As for C2 , the departing NO2moiety supplies little to the HOMO but it has a large contribution in to the LUMO, so that the LUMO is entirely on it. In the case of D two levels of calculations yield different patterns for each of these orbitals (see the
Figure 6).


Fig.6 The frontier molecular orbitals of nTNAZ+Zn composites (n∶1,2)
Figure 7 stands for UV-VIS spectra (time-dependent DFT) of the composites considered.

Fig.7 UV-VIS spectra of TNAZ, 2TNAZ and the composites considered (B3LYP/6- 31++G(d,p))
As compared to C1 spectrum, C2 spectrum exhibits a hypsochromic effect. The dimeric arrangement of TNAZ molecules around Zn atom causes effective bathochromic effect to happen in the case of 2TNAZ+Zn. As seen in the
Figure, Zn atom highly affects the spectra of TNAZ and 2TNAZ systems.
Figure 8 displays the electrostatic potential maps of composites C1, C2 (more stable conformer of TNAZ+Zn composite) and D. Where, the red/reddish and blue/bluish regions stand for the negative and positive potential fields, respectively. In composite D, the electrostatic potential field over TNAZ components are unequal, especially the regions of C—NO2and N—NO2moieties compared componentwise are highly different. This effect should arise from the unequal sharing of electron population donated by the zinc atom to each TNAZ component.

Fig.8 Electrostatic potential map of two of the composites (B3LYP/6-31++G(d,p))
3 Conclusion
Within the restrictions of the density functional theory, calculations at the levels of B3LYP/6-31++G(d,p) andωB97X-D/6-31++G(D,P) have been performed in order to investigate the effect of zinc on TNAZ at the molecular level. The following main results are obtained.
(1)Of the two C—NO2and one N—NO2bonds, only one of the C—NO2bond is affected by zinc atom in TNAZ+Zn composite.
(2)The NO2moiety expelled from TNAZ and originating from the broken C—NO2bond has negative over all charge.
(3)It has been found that as the zinc percent decreases the composite system seems to get some stability to keep the C—NO2and the other bonds in reasonable lengths.
(4)As the zinc percent decreases the composite system becomes more susceptible to impact stimulus.
(5) Of course, decreasing the zinc content eliminates any beneficial thermal output if it is expected from the composite formulation.
