
NCSTN基因沉默对HaCaT细胞增殖分化的影响
2020-10-24 04:21张婉璐张媛媛吴英达程萍李文锐徐浩翔王宝玺何艳艳李诚让
中华皮肤科杂志 2020年9期
张婉璐 张媛媛 吴英达 程萍 李文锐徐浩翔 王宝玺 何艳艳 李诚让
1中国医学科学院北京协和医学院皮肤病医院皮肤科,南京210042;2中国医学科学院北京协和医学院整形外科医院皮肤科,北京100144
张婉璐现在蚌埠医学院第一附属医院皮肤科,安徽233004
反常性痤疮(acne inversa,AI)的发病与遗传、环境、免疫、肥胖、吸烟等多种因素有关[1],具体机制仍不清楚。2010年,Wang等[2]首次发现,部分家族性AI 的发病与编码γ 分泌酶不同亚单位的基因功能性突变有关。Nicastrin(NCT)蛋白参与γ分泌酶的组装、成熟及稳定,已发现的AI患者基因突变中,NCSTN 基因突变占大多数[3],常引起AI 患者NCT 蛋白表达下调[4]。γ 分泌酶切割Notch 受体蛋白参与Notch 信号通路,而Notch 信号通路在表皮细胞的分化、增殖,毛囊的终末分化中发挥关键作用[5]。目前认为AI 的始动因素是毛囊漏斗部的角化过度及栓塞[6⁃7],组织学上毛囊漏斗部的上皮形态及角化方式与表皮中的角质形成细胞类似。细胞角蛋白(cytokeratin,CK)、内披蛋白(involucrin,IVL)和兜甲蛋白(loricrin,LOR)都是角质形成细胞分化的标志[8]。本研究通过检测NCSTN 基因沉默的人永生化角质形成细胞(HaCaT 细胞)的增殖活性及多种角蛋白和其他分化分子(IVL、LOR)的表达,探究NCSTN 基因对角质形成细胞增殖分化的影响,为后续探究NCSTN 基因突变引起反常性痤疮提供新思路。
材料与方法
一、主要试剂与材料
HaCaT 细胞为本实验室保存。高糖型DMEM培养基、胎牛血清和胰酶(美国Gibco公司),慢病毒载体、质粒、RNAi⁃Mate 转染试剂(上海吉玛制药技术有限公司),Trizol 试剂(美国Invitrogen 公司),PrimeScriptTMRT⁃PCR 反转录试剂盒(日本TaKaRa公司),实时定量PCR 试剂盒AceQ®qPCR SYBR®Green Master Mix(Without ROX Premixed)(南京诺唯赞生物科技股份有限公司),RIPA 裂解液(强)、cocktail蛋白酶抑制剂、BCA蛋白浓度测定试剂盒、Western 印迹转膜液、电泳液(上海碧云天生物技术有限公司),牛血清白蛋白(南京生兴生物技术有限公司)、Tris⁃Glycine eXtended(TGX)预制胶4%~15%,Clarity ECL化学发光液(美国Bio⁃Rad公司),CCK8 试剂盒(日本同仁化学研究所),兔抗人NCT 多克隆抗体、兔抗人IVL 多克隆抗体、兔抗人LOR多克隆抗体(美国Proteintech公司),辣根过氧化物酶标记的兔抗人β肌动蛋白单克隆抗体(美国Cell Signaling 公司),兔抗人CK1多克隆抗体、兔抗人CK5/10/16/19单克隆抗体,辣根过氧化物酶标记的山羊抗兔IgG H&L抗体(英国Abcam公司)。
二、方法
1.构建慢病毒介导shRNA沉默NCSTN基因的HaCaT 细胞模型:参考文献[9]并在相同实验条件下,用慢病毒介导的靶向NCSTN 基因特异性的短发卡RNA(shRNA)表达质粒转染HaCaT细胞,构建慢病毒介导shRNA沉默NCSTN基因的HaCaT细胞模型。正常培养的HaCaT细胞分为空白组(仅正常培养)、阴性对照组(转染空载慢病毒)和shRNA 组(转染靶向NCSTN基因的慢病毒)。流式细胞仪验证转染效率,并经实时定量PCR 及Western 印迹法验证基因沉默效率,实验重复3次。
2. 实时定量PCR 检测HaCaT 细胞中NCSTN、角蛋白、LOR及IVL mRNA的表达:按Trizol说明书提取shRNA 组及阴性对照组细胞总RNA,超微量分光光度计测定RNA 浓度,随后使用PrimeScriptTMRT⁃PCR 试剂盒将RNA 反转录为cDNA,并用超微量分光光度计测定DNA浓度。检索NCBI GenBank数据库设计引物,由生工生物工程(上海)股份有限公司负责合成,PCR 引物序列见表1。使用AceQ®qPCR SYBR®Green Master Mix 试剂盒,按说明书进行实时定量PCR。以β 肌动蛋白为内参,采用2-ΔΔCt值表示目的基因mRNA 相对表达量,△Ct = Ct目的基因-Ctβ肌动蛋白,△△Ct = △CtshRNA组-△Ct阴性对照组。实验重复3次。

反向引物GCGATGTAATGTTGAAGAGGC CTCTGCATTTGTCCGCTTGT CCAGAGGAAACACTGCTTGTGA ATATTCACGGCTCCCACTCC GCGCAGAGCTACCTCATTCT CCATGACCTTGGTGCGGATT ACCTCGCGGGAAGAATAGGA ACAATGGTACGCACCTGACG GGTCTCCTTCTCGTTCTGGATG CTGGGCTTCAATACCGCTGA GCGTCTCAGCTCCGTTAGTT TGGGGTTGGGAGGTAGTTGT CAGCAGTCATGTGCTTTTCCT GCTGTCACCTTCACCGTTCC基因名称NCSTN CK1 CK5 CK7 CK10 CK14 CK16 CK17 CK18 CK19 CK20 LOR IVL β肌动蛋白正向引物AATGTGAGCTATCCCGAATG CCGAAGGAGAGTGGACCAAC TCAAGAAACAGTGCGCCAAT GCAGGCTGAGATCGACAACA ATGGCAACTCACATCAGGGG GCAGCAGAACCAGGAGTACA CTACGCTGTGAGATGGAGCA GCTCAGCATGAAAGCATCCC TGAGTCCTGTCCTTTCTCTCTC GAAGGATGCTGAAGCCTGGT TGGCCTACACAAGCATCTGG AGCTCTCATGATGCTACCCG TCCTCCAGTCAATACCCATCAG CCATCGTCCACCGCAAAT
3. Western 印迹法检测HaCaT 细胞中NCT、角蛋白、LOR及IVL蛋白的表达:采用RIPA裂解液及cocktail蛋白酶抑制剂的混合物提取shRNA组及阴性对照组细胞总蛋白,BCA法测定蛋白浓度。配制10% SDS 聚丙烯酰胺凝胶,每孔加入60 μg 蛋白样品。使用湿转法电转至PVDF 膜,5%牛血清白蛋白封闭2 h,分别加入NCT、IVL、LOR、CK1、β 肌动蛋白抗体,1∶1 000 稀释;CK5、CK10、CK16 抗体,1∶10 000 稀释;CK19 抗体,1∶50 000 稀释。4 ℃过夜;再加入1∶2 000稀释的山羊抗兔二抗,室温孵育2 h,TBST 洗膜5 次,每次6 min,ECL 发光。Image J 软件测定相关分化分子蛋白灰度值。待测蛋白相对表达量=待测蛋白条带灰度值/β 肌动蛋白条带灰度值。实验重复3次。
4.CCK8 实验检测HaCaT 细胞的增殖活性:取shRNA 组及阴性对照组对数生长期细胞,加入含10%胎牛血清的完全培养基,制成(5 ~10)×104个/ml细胞悬液。向96孔板各孔中加入100 μl细胞悬液,设置12、24、36、48、60和72 h时间点,每个时间点设置5个复孔,放入37 ℃、5%CO2培养箱中培养。每孔加入10 μl CCK8溶液,37 ℃孵育1 h,测定各时间点450 nm处的吸光度(A值),代表其增殖能力。
5. 统计学分析:应用GraphPad Prism V 7.0 和SPSS 23.0 软件进行统计学分析并绘制统计图,数据用±s 表示。两组计量资料的比较采用两独立样本t检验,P<0.05为差异有统计学意义。
结果
一、HaCaT 细胞慢病毒转染效率及NCSTN基因沉默效率的测定
慢病毒转染后第5 天,空白组HaCaT 细胞形态为上皮样细胞贴壁生长;阴性对照组和shRNA 组细胞生长状态良好,形态一致,大小均匀,呈多角形,贴壁生长,排列紧密。倒置荧光显微镜下(图1),阴性对照组和shRNA 组可见绿色荧光,空白组未见荧光表达,提示慢病毒载体系统能有效转染HaCaT 细胞。流式细胞仪检测显示,阴性对照组和shRNA 组稳定转染HaCaT 细胞荧光率均为90%以上,见图2。
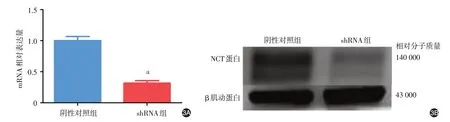
实时定量PCR 及Western 印迹结果显示,shRNA 组NCSTN mRNA 及蛋白表达(0.42 ± 0.19、0.30 ± 0.07)均显著低于阴性对照组(1.00 ± 0.34、1.00 ± 0.26;t = 5.196、2.637,P < 0.001、< 0.05),见图3。与阴性对照组相比,shRNA 组蛋白表达量下降了70%左右,因此沉默效率达70%。
二、稳定沉默NCSTN 基因对HaCaT 细胞增殖活性的影响
CCK8法显示,转染慢病毒24 ~72 h,shRNA组细胞增殖活性均高于阴性对照组,见图4。

三、实时定量PCR检测各组细胞中不同角蛋白及IVL、LOR mRNA的表达
如图5 所示,与阴性对照组相比,shRNA 组HaCaT 细胞中角蛋白CK5、CK1、CK10 mRNA 表达显著升高(t = 4.330、5.407、3.971,P < 0.05、<0.001、<0.01),CK16、CK19、IVL mRNA 表达显著降低(t = 5.005、4.933、9.114,P < 0.001、< 0.01、<0.001)。两组间CK7、CK14、CK17、CK18、CK20及分化分子LOR表达差异均无统计学意义(t=1.454、1.657、0.175、1.230、0.342、1.275,均P>0.05)。
四、Western 印迹法检测干扰NCSTN 基因表达后对分化分子蛋白表达的影响
如图6 所示,干扰NCSTN 基因表达后,shRNA组CK5、CK1、CK10、LOR蛋白相对表达与阴性对照组相比差异无统计学意义(t=0.247、0.317、0.343、0.016,均P>0.05),而CK16、CK19和IVL蛋白表达显著低于阴性对照组(t = 3.787、3.817、2.904,P <0.01、<0.05、<0.05)。
讨论

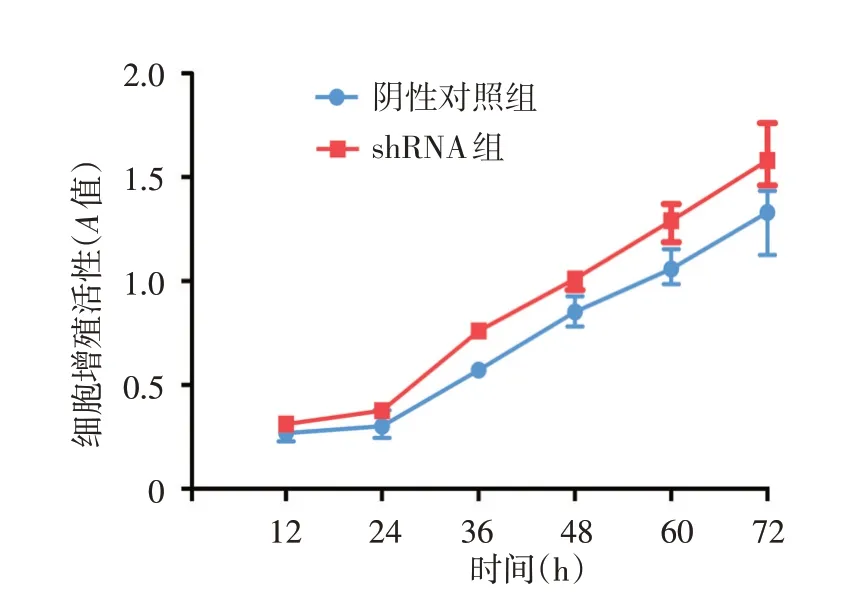


AI 的早期损害表现为毛囊漏斗部上皮的角化过度及异常增生、表皮囊肿形成[6⁃7]。本文CCK8实验显示,NCSTN 基因稳定沉默的HaCaT 细胞增殖活性明显增强,这或许可以解释AI 皮损中表皮银屑病样增生模式[10]。我们还发现,NCSTN 基因稳定沉默的HaCaT 细胞中,角蛋白CK16、CK19 及终末分化分子IVL表达均显著降低。
角蛋白是上皮细胞主要的结构蛋白和分化的标志产物,在细胞内外信息传导、基因调控方面发挥重要作用,其表达模式多种多样。皮肤基底层中未分化的角质形成细胞主要表达CK5和CK14,而分化特异性角蛋白CK1和CK10表达于角质形成细胞分化的初始阶段,是表皮的主要结构蛋白,其中CK1位于基底上层,表达于即将角化的上皮细胞内,CK10主要表达于角化的复层鳞状上皮中处于角化前期的基底上层细胞内[11]。本研究结果显示,shRNA组HaCaT细胞中CK1 mRNA表达明显高于阴性对照组,但其蛋白表达与阴性对照组差异无统计学意义。由于基因表达的调控层次很多,转录水平的调控只是一个环节,转录后调控、翻译及翻译后调控均对蛋白的表达有一定影响,因此,基因的mRNA丰度不一定与其翻译产物蛋白质的表达呈线性关系[12]。此外,NCSTN基因稳定沉默后HaCaT细胞中早期分化分子CK1蛋白表达并无明显变化,也提示NCSTN突变可能不影响角质形成细胞的早期分化。
生理情况下,CK16在表皮无表达,但在表皮过度增生性疾病如银屑病中,CK16 可大量表达[13]。因此,CK16可作为细胞增殖分化的标志分子之一,其表达属于细胞过度增殖相关联的替代分化途径[14]。Janse等[15]认为,CK16表达上调可能是AI发病的继发事件,先天免疫系统的激活也可能导致CK16 在皮损内表达上调。但本研究结果显示,NCSTN 基因稳定沉默的HaCaT 细胞中CK16 表达明显下调。临床上KRT16 基因突变可导致先天性厚甲综合征,出现厚甲伴掌跖角化。KRT16基因突变导致CK16 蛋白螺旋结构发生改变,并在细胞中异常聚集,CK16蛋白的正常组装受到干扰,导致异常角蛋白形成[16]。KRT16 基因敲除或部分敲除的小鼠模型出现掌跖角化样的皮肤损害,也提示CK16 蛋白异常表达可能导致角化过度[17]。因此,我们推测NCSTN 基因沉默引起的CK16 蛋白表达异常也可能介导表皮角化过度。
CK19 是皮肤、毛囊等处表皮干细胞的标志蛋白[18],表皮干细胞是皮肤组织特异性的、具有自我更新、高度增殖能力和多向分化潜能的一类细胞,定位于表皮的基底层及毛囊隆突部[19]。生理情况下,CK19仅分布于表皮基底层、外毛根鞘、皮脂腺和汗腺,当表皮干细胞分化成为成熟角质形成细胞后,CK19表达下降,因此可通过测定CK19表达来反映表皮干细胞的分化能力[20]。研究[21]表明,当组织创伤修护时,CK19表达增加,提示表皮干细胞在分化的同时可能也在自我复制。本研究结果显示,NCSTN基因稳定沉默的HaCaT细胞中CK19表达下调,提示表皮干细胞分化能力降低,可能同时伴随增殖能力减弱,导致AI皮损的自我修复能力降低。CK19在AI发病中可能发挥作用,但其作用仍不明确。
IVL 是一种可溶性胞质蛋白,主要表达于正常表皮棘细胞上层和颗粒层,目前IVL多作为研究皮肤角质形成细胞终末分化的标志蛋白[22]。本研究显示,NCSTN基因沉默的HaCaT细胞IVL蛋白表达明显下降,提示表皮角质形成细胞的终末分化可能延迟。
综上所述,本研究显示,NCSTN 基因沉默的HaCaT 明显过度增殖,同时角蛋白CK16、CK19 及分化分子IVL 表达显著下降。结合相关文献我们推测,NCSTN 功能缺失可能部分导致毛囊漏斗部异常角化,终末分化延迟,AI皮损处角质形成细胞过度增殖,毛囊角栓形成,从而导致膨胀的毛囊漏斗部破裂释放内容物,继发严重的炎症反应及感染,最终导致AI 的发生。由于疾病的发生发展是一个复杂的过程,而我们仅在NCSTN 基因沉默的HaCaT细胞中进行了初步探究,NCSTN基因突变是否导致免疫细胞发生改变,动物实验及人类组织是否有相似表现仍需要进一步深究。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
纺织报告(2022年5期)2022-11-22
中国畜牧杂志(2022年1期)2022-11-06
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
毛纺科技(2021年3期)2021-04-06
中国麻风皮肤病杂志(2021年2期)2021-01-02
儿童故事画报·智力大王(2020年1期)2020-04-28
凤凰生活(2018年6期)2018-06-25
东方教育(2017年14期)2017-09-25
课程教育研究·学法教法研究(2016年1期)2016-03-17
