
基于16S rRNA基因测序的寻常型银屑病患者肠道菌群差异研究
2020-10-24 04:21王丽玮徐浩翔段志敏崔盘根龚春燕李岷
中华皮肤科杂志 2020年9期
王丽玮 徐浩翔 段志敏 崔盘根 龚春燕 李岷
中国医学科学院北京协和医学院皮肤病医院皮肤科,南京210042
银屑病是一种免疫介导的慢性炎症性皮肤病,寻常型银屑病最为常见[1]。银屑病的发病与多种因素有关。肠道菌群是生活在人类消化道中的复杂微生物群落[2],被证实与肥胖、代谢综合征[3]、类风湿性关节炎[4]等自身免疫性疾病有关。有研究报道肠道菌群与银屑病密切相关[5⁃7],本文探究江苏地区成年寻常型银屑病患者与健康人肠道菌群的差异。
对象与方法
一、对象
2017 年9 月至2018 年2 月中国医学科学院皮肤病医院住院治疗的银屑病患者,以本院健康体检的员工及周边居民为健康对照。依据中华医学会《临床诊疗指南:皮肤病与性病分册》[8]指导诊断,并完成银屑病面积和严重程度指数(psoriasis area and severity index,PASI)评分。纳入标准:①银屑病患者及健康人均需为南京市及周边县市常住居民,近1年无外地久居经历;②未合并消化道出血、糖尿病、高血压等其他已知影响肠道菌群的系统性疾病;③1 个月内无痔疮发作、腹泻等影响粪便形态及主要成分的事件;④1 个月未使用抗生素、活菌类制剂;⑤未在产褥期、哺乳期及妊娠期;⑥受试者配合采集标本前1周不使用类固醇制剂、免疫抑制剂、中草药及其他可能影响消化道菌群的制剂,不服用酸奶、米酒及其他活菌类食品。本研究通过中国医学科学院皮肤病医院医学伦理委员会批准[批号:(2017)临审第(022)号],所有受试者均签署知情同意书。
二、粪便标本采集
取无菌袋采集受试者的新鲜粪便,立即放入冰盒后转移入实验室取样,2 h 内分装于1.5 ml 无菌离心管,置-80 ℃冰箱冻存,干冰冻存运送至苏州协云基因科技有限公司实验室。
三、DNA提取与扩增及16S rRNA基因测序
依据粪便基因组DNA 提取试剂盒[DP328,天根生化科技(北京)有限公司]提取样本DNA。DNA 样本经Qubit2.0 荧光定量(美国Invitrogengon公司)验证后,选择16S rRNA的V3-V4高变区对应基因序列作为扩增和测序的目的片段。引物由苏州金唯智生物科技有限公司合成,341 正向引物:5′⁃CCTAYGGGRBGCASCAG⁃3′;806 反向引物:5′⁃GGACTACHVGGGTWTCTAAT⁃3′。使用PCR 试剂盒(美国Biosystems 公司)扩增,扩增条件为:95 ℃预变性3 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,25个循环;72 ℃延伸5 min。PCR产物利用琼脂糖凝胶电泳,选择465 bp 长度条带,割胶并回收纯化目标条带。
将扩增溶液等浓度导入测序仪(美国Illumina公司),采用双端2×300策略测序,并利用MiSeq控制软件进行图像分析和数据转化。
四、数据分析
对获得的原始Reads 进行拼接、质控后得到Tags,使用Usearch 软件(Version 8.1.1861),基于基因组在线数据库(Gold)过滤嵌合体序列,获得有效Tags 后,使用QIIME 软件(Version 1.9.1)按照大于97% 的相似性聚类为一个分类操作单元(operational taxonomic units,OTU)。
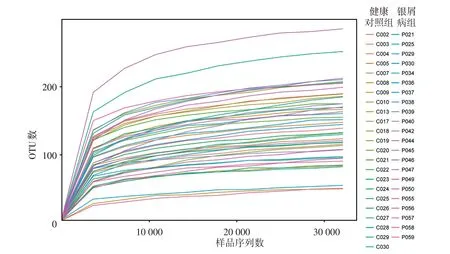
1.稀释曲线构建:以样品测序量为横坐标,对应的OTU 数目为纵坐标构建稀释曲线,稀释曲线可直接反映测序数据量的合理性,并间接反映样品中物种的丰富程度。当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量只会产生少量新OTU;反之表明不饱和,增加数据量可以发现更多OTU。
2.α 多样性分析:α 多样性指生境群落内微生物群落的丰度和多样性;采用QIIME软件计算OTU数目及α 多样性主要指数Chao1 指数、Shannon 指数和Simpson 指数。Shannon 指数用于估算样品中微生物多样性,指数值越高,表明群落的多样性越高;Chao1 指数用于估计物种总数,值越大代表物种总数越多;Simpson指数值越大,说明群落多样性越低。
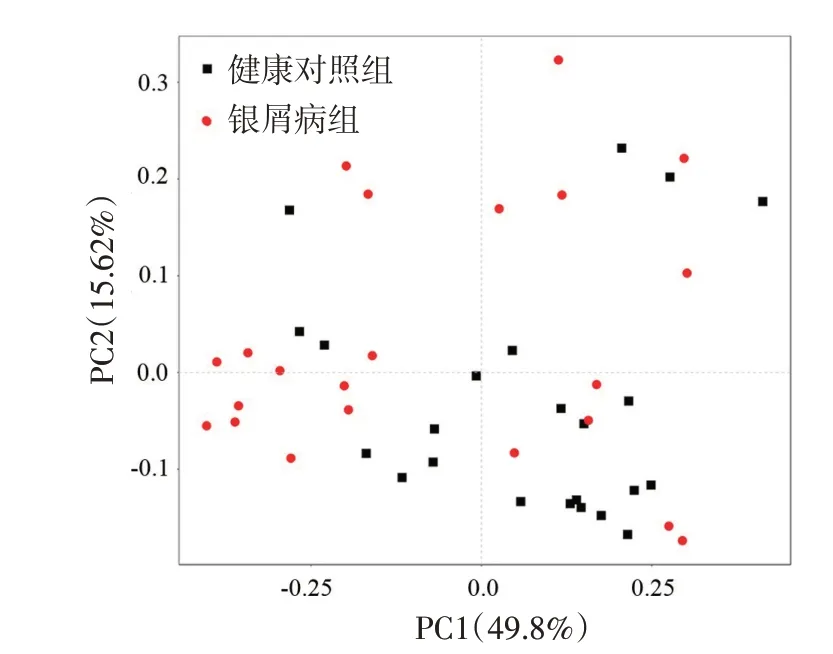
3.β 多样性分析:β 多样性指生境群落间微生物群落的差异;PCoA 分析可从复杂的多维变量数据中提取主要变量行降维至2 个变量并建立主要坐标,对整体样本进行简化的数据描述;以对组间差异贡献最大的两个变量值(第一主成分PC1、第二主成分PC2)分别作为横坐标及纵坐标定位每一样品,并分别注释主成分对整体组间差异的解释度;采用置换多元方差分析两组群落组成结构差异,P<0.05为差异有统计学意义。
4. 菌群分布及差异分析:对照Silva 数据库(Release 119)进行物种注释和分类,采用秩和检验分析各层级样本物种差异。采取线性判别分析效应量(linear discriminant analysis Effect Size,LEfSe)分析[9]确定最有可能解释类别之间差异的特征物种,对数据进行降维、评估差异显著的物种的线性判别分析(linear discriminant analysis,LDA)积分,定义LDA 评分大于2 且具有统计学差异的物种可以作为生物标志物;LEfSe 分析结果以条形图展示LDA评分结果,以进化分枝树图展示微生物标志物的物种之间的进化关系;以总丰度为1计算厚壁菌门及拟杆菌门相对丰度并计算其比值,以中位数(P25,P75)表示。
五、统计学方法
采用SPSS 19.0 软件,通过Kolmogorov⁃Smirov检验变量是否服从正态分布,正态分布样本资料以±s 表示,采用Welch′s t 检验,非正态分布样本采用秩和检验。P<0.05为差异有统计学意义。
结果
一、临床资料
寻常型银屑病患者22 例,男16 例,女6 例,年龄19 ~65岁;健康对照组23例,男17例,女6例,年龄25 ~60 岁;两组之间性别构成比、年龄、体质量指数(BMI)差异均无统计学意义(P>0.05)。见表1。银屑病组中2例女性、4例男性静脉血尿酸值增高,1例男性十余年前因外伤切除脾脏,余患者及健康对照均无系统疾病。所有受试者均无银屑病家族史。
二、α多样性分析
稀释曲线显示(图1),随着样品测序量的增大,检测到的物种种类随之增加,在测序至30 000序列以上后,新增OTU的数量均趋于稳定。

组别银屑病组健康对照组t值P值例数22 23年龄(岁)37.32±15.59 36.87±11.86 0.11 0.914 BMI(kg/m2)23.88±3.21 22.78±2.72 1.24 0.221
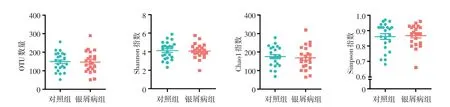
银屑病组OTU 数目(147.55±57.07)与健康对照组(148.96±50.45)比较,差异无统计学意义(t=0.09,P = 0.930);银屑病组Shannon 指数(4.08 ±0.80)、Chao1 指数(169.52 ± 63.17)、Simpson 指数(0.87 ± 0.07)与健康对照组(分别为4.11 ± 0.94、175.36±53.59、0.86±0.90)比较,差异均无统计学意义(t=0.12、0.34、0.27,均P>0.05)。见图2。
三、β多样性分析
PCoA分析显示(图3),银屑病组与健康对照组的第一主成分解释度为49.8%,第二主成分解释度为15.62%。置换多元方差分析显示,两组群落组成结构差异有统计学意义(P=0.011)。
四、菌群分布及差异分析
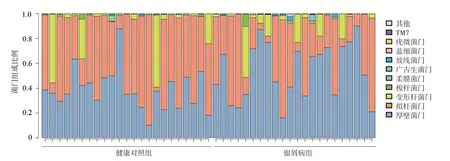
1.菌门的相对丰度分析:两组共45个样品中,占主要比例的菌门相对一致,为厚壁菌门、拟杆菌门、变形杆菌门、梭杆菌门;不同样本的菌门组成差异较大。见图4。
2.LEfSe 分析:银屑病组显著升高且具有生物标志物作用的物种有厚壁菌门、梭菌纲、梭菌目,丹毒丝菌目、丹毒丝菌科;健康对照组显著升高的生物标志物物种有艾普西隆变形杆菌纲、弯曲杆菌目、弯曲杆菌科、弯曲杆菌属,拟杆菌目、拟杆菌科(图5A)。
具有微生物标志物的物种进化关系如下:厚壁菌门-梭菌纲-梭菌目,厚壁菌门-丹毒丝菌所属纲-丹毒丝菌目-丹毒丝菌科,变形杆菌门-艾普西隆变形杆菌纲-弯曲杆菌目-弯曲杆菌科-弯曲杆菌属,拟杆菌门-拟杆菌纲-拟杆菌目-拟杆菌科(图5B)。
3.高尿酸血症对银屑病患者的影响:6 例伴高尿酸血症与16 例无高尿酸血症患者的年龄、BMI、PASI 评分差异无统计学意义(P > 0.05),见表2。两组间厚壁菌门、拟杆菌门的相对丰度及其比值差异均无统计学意义(P>0.05)。

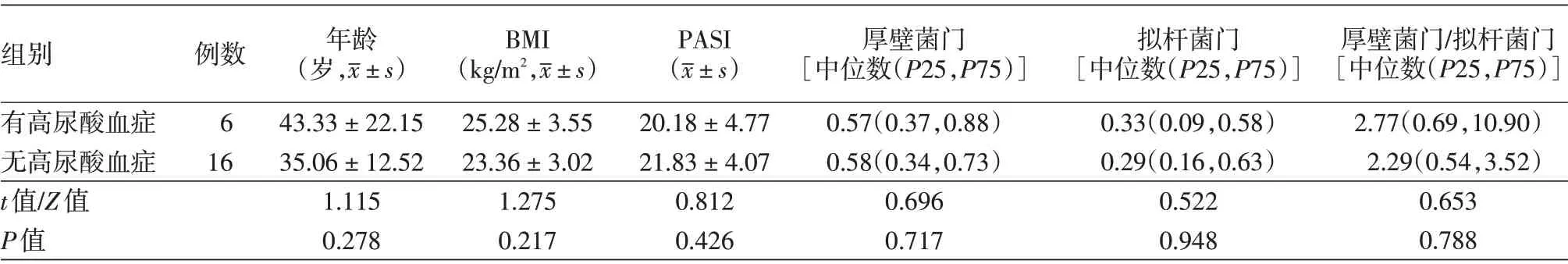
例数6 16年龄(岁,images/BZ_37_1351_1170_1373_1213.png±s)BMI(kg/m2,images/BZ_37_1351_1170_1373_1213.png±s)组别有高尿酸血症无高尿酸血症t值/Z值P值PASI(images/BZ_37_1351_1170_1373_1213.png±s)43.33±22.15 35.06±12.52 1.115 0.278 25.28±3.55 23.36±3.02 1.275 0.217 20.18±4.77 21.83±4.07 0.812 0.426厚壁菌门[中位数(P25,P75)]0.57(0.37,0.88)0.58(0.34,0.73)0.696 0.717拟杆菌门[中位数(P25,P75)]0.33(0.09,0.58)0.29(0.16,0.63)0.522 0.948厚壁菌门/拟杆菌门[中位数(P25,P75)]2.77(0.69,10.90)2.29(0.54,3.52)0.653 0.788
讨论
本研究利用16S rRNA基因测序技术分析22例成年寻常型银屑病与23例成年健康对照组的肠道细菌群,研究纳入的银屑病患者与健康对照之间的性别分布相似,年龄及BMI 差异无统计学意义,具有可比性。有学者曾指出[10],不同国家的个体之间粪便微生物群的系统发育组成存在显著差异,在同一种族中,细菌群落的系统发育组成在出生后3年内逐渐演变成成人样构型,儿童之间的人际差异明显大于成人,因此本研究只纳入汉族成人样本;在样本采集中充分考虑了地域饮食习惯、治疗方案、影响肠道菌群的慢性疾病、下消化道疾病、短期内影响肠道菌群的饮食因素,采取无菌袋收样,与国际上研究人源肠道菌群的标本采集原则一致[11]。
有研究发现,在关节型银屑病及寻常型银屑病中均可观察到肠道细菌群的多样性较健康人显著下降[12],这一结论在其他研究中并未能重复,部分研究中银屑病患者的肠道菌群α 多样性可呈现出下降趋势[5⁃7]。本研究发现,寻常型银屑病患者与健康人肠道菌群α多样性无差别,但菌群总数较健康人有显著降低,这与部分既往研究一致[7,13]。目前关于α多样性和菌群总量的研究结果并不一致,这种差异可能是由于研究对象、采样条件与研究技术的不同造成。在PCoA分析中,样品距离越远,表示物种组成结构差异越大,本研究中银屑病组与健康对照组第一主成分与第二主成分解释度之和>50%,置换多元方差分析两组间β 多样性差异显著,提示银屑病与健康人的肠道菌群具有显著的组间菌群差异,表明银屑病患者的肠道菌群较健康人群总体发生了可以被检测出的变化,且这种变化趋势为菌群总量或多样性的降低。
关于丰度差异的具体菌群,本研究发现,在银屑病组中显著升高的有厚壁菌门、梭菌纲、梭菌目,丹毒丝菌目、丹毒丝菌科;在健康对照组显著升高的物种有艾普西隆变形杆菌纲、弯曲杆菌目、弯曲杆菌科、弯曲杆菌属,拟杆菌目、拟杆菌科。Scher等[12]通过16S rRNA基因高通量测序及微生物数据库比对分析发现,31 例银屑病患者的粪便样本中粪球菌属种类减少。我国研究[6]发现,银屑病患者粪便标本中阿克曼氏菌的丰度较健康人显著降低;另一项研究中[7],银屑病患者粪便中普拉氏梭杆菌丰度较健康对照组显著降低,而大肠杆菌丰度显著增高。由于人群的局限性及肠道微生物组受人种、地域、性别、年龄等多种因素影响,目前临床队列研究数量及规模有限,对肠道细菌群的具体变化无法得出相对一致的结论。
但值得注意的是,本研究对于银屑病及健康对照组中的差异菌群的统计方法与目前大多数类似的研究不同,已发表的文献多采用单因素方差分析或非参数Kruskal⁃Wallis 秩和检验分析组间差异,本研究加入LEfSe 分析[14],可充分考虑组内方差最小、组间方差最大的选择。本研究通过菌种相对丰度排序得知,样品间同一物种的相对丰度有较大差异,但相对丰度的优势物种一致,厚壁菌门和拟杆菌门均为肠道菌群的主要菌群,与其他相关研究的结论一致[2],通过LEfSe分析发现,银屑病组显著升高的具有生物标志物作用的物种为厚壁菌门。有研究提出,微生物可能通过多种方式促进代谢,包括修饰胆汁盐及其产生来提高脂肪酸的生物利用度[15],直接影响宿主的脂解活性,导致脂肪酸氧化速率降低,从而使更多的脂肪酸吸收[16]。厚壁菌门与肥胖密切相关,肥胖人群肠道菌群中拟杆菌门降低,厚壁菌门升高,在经历饮食治疗体重降低后,拟杆菌门的相对丰度增加,而厚壁菌门的丰度减少,并与饮食类型无关。在银屑病患者中以肥胖、高尿酸血症等代谢紊乱为临床表现的代谢综合征为常见的并发症,是否因肠道菌群的改变导致或促进了银屑病患者中代谢综合征的发生还需更多证据。在本研究中伴与不伴发高尿酸血症的银屑病患者之间,BMI、PASI评分及厚壁菌门、拟杆菌门的相对丰度及比例差异均无统计学意义,肠道菌群与银屑病患者中代谢综合征的关系还需进一步人群随访及动物实验验证。
已有研究通过动物模型证明,干涉饮食及影响肠道菌群可影响疾病的发生与发展[17],粪便移植也逐步用于疾病的治疗[18],我们的研究为进一步研究粪便移植或饮食干预治疗银屑病提供了可能的方案。本文的不足之处在于样本量有限,虽严格控制变量,但研究对象局限于江苏地区的健康人及银屑病患者。目前银屑病患者与健康人肠道菌群的构成及相对丰度在不同研究中存在较大的差异,可能与研究对象、检测方法、技术差异与样本量的限制有关,进一步研究肠道菌群与银屑病的关系,有利于为探究银屑病的发病机制及治疗提供更多依据。
志谢苏州协云基因科技有限公司
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
世界中医药(2022年22期)2022-12-14
中国特种设备安全(2022年2期)2022-07-08
汽车实用技术(2022年11期)2022-06-20
中国农学通报(2022年14期)2022-06-01
油气田环境保护(2022年2期)2022-05-09
皮肤病与性病(2021年3期)2021-07-30
山东畜牧兽医(2021年5期)2021-06-07
中国沼气(2019年1期)2019-04-13
世界家苑(2018年8期)2018-09-04
山东工业技术(2016年15期)2016-12-01
