
丙氨酸Mg2+配合物的手性转变机理水分子(簇)的作用及水溶剂效应*
2020-10-20 09:39:20张雪娇刘芳吴梓昊徐锐英马宏源杨晓翠王佐成
中山大学学报(自然科学版)(中英文) 2020年5期
张雪娇,刘芳,吴梓昊,徐锐英,马宏源,杨晓翠,王佐成
(1. 白城师范学院理论计算中心,吉林白城137000;2. 白城师范学院传媒学院,吉林白城137000;3. 白城师范学院物理学院,吉林白城137000;4. 中山大学环境学院,广东广州510257)
丙氨酸(alanine, Ala)有α−和β−丙氨酸两种,本工作讨论的是手性α−丙氨酸。根据旋光性可分为左−丙氨酸(L−Ala) 和右−丙氨酸(D−Ala),根据构型可分为S−丙氨酸(S−Ala)和R−丙氨酸(R−Ala)。不同旋光性的Ala 具有不同的作用,生命体内L−Ala 是组成蛋白质的重要成分;通常生命体内只有微量的D−Ala,可阻止脂质氧化损伤,清除细胞毒性,促进细菌孢子代谢;过量的D−Ala 会导致某些疾病或衰老,生命体内D−Ala 来源之一可能是L−Ala的消旋[1−4],因此L−Ala消旋的研究尤为重要。
基于上述原因,学者们在Ala 的手性转变领域做了大量的工作。文献[5]的实验研究发现,光学纯Ala 分子的手性是可以改变的。文献[6−9]用DFT 理论研究了气相单体Ala 的手性转变及水分子(簇)的作用,结果表明:Ala 分子消旋反应优势通道的表观能垒大约是260.0 kJ·mol−1;水分子(簇)的催化可使其缓慢消旋。文献[10]在ONI⁃OM (CAM−B3LYP/ 6−31+G (d, p): UFF) 水平,研究了SWBNNT 对Ala 旋光异构的限域影响,结果表明:SWBNNT(5,5)对Ala 的消旋具有明显的限域催化作用。文献[11]的密度泛函理论研究表明:水液相下中性的Ala 分子可以缓慢地消旋,氢氧根水分子簇的催化能使消旋速度大幅增加。文献[12]的DFT 理论研究表明:水液相下Ala 两性离子可以极缓慢地的实现手性转变。文献[13]用DFT理论了气相的Cu2+与Ala配合物的手性转变,结果表明,Cu2+对Ala 的消旋反应具有催化作用。文献[14] 用DFT 理论研究了气相丙氨酸Ca2+配合物的手性转变及水分子的催化,结果表明:Ca2+对Ala 的手性转变具有催化作用,水分子对丙氨酸Ca2+配合物的手性转变具有较好的催化作用。
Mg2+是人体内丰富的阳离子,与氨基酸螯合后参与蛋白质的合成和多种酶促反应[15−16],研究气相的“金属离子-生物分子”体系,可获得相关体系最本质的物理化学性质等信息[17−19],学者们对气相的“Mg2+与氨基酸的作用”做了大量的工作[17−21],结果表明,氨基酸Mg2+螯合物具有很高的结合能,构象稳定,比较而言,两性氨基酸Mg2+螯合物的稳定性好于中性氨基酸Mg2+螯合物。光学纯的手性分子的优构体对生命体有积极的作用,而其对映体则无用甚至有害,因此,光学纯的手性分子是否容易消旋对生命的健康极为重要。然而,关于氨基酸Mg2+螯合物手性转变的研究鲜见报道,Ala 是重要的手性氨基酸,基于此考虑到水分子(簇)的普遍存在以及生命体内的水溶剂环境,结合以往的研究经验[6−14],本工作采用密度泛函理论对标题反应进行了研究。
1 研究与计算方法
采用处理含金属和弱作用体系具有较高精度的杂化泛函M06 方法[22],在6−31++G(d, p)[23]基组下优化反应过程中单重态势能面上的驻点结构,同时获得吉布斯自由能热校正(计算表明本工作研究的体系在可能的自旋态1、3 和5 中,单重态最稳定);采用自然键轨道NBO 方法计算了相关体系的NPA 电荷;通过对过渡态[24]进行IRC(内禀反应坐标)计算[25],对过渡态的可靠性进行确认。为得到较高精度的反应过程势能面并兼顾计算成本,在M06/6−311++G(2df, pd)高角动量基组水平下计算单点能。水溶剂效应结合自洽反应场理论的SMD 模型[26]方法处理。总吉布斯自由能用Gtotal=Gtc+ESP计算(Gtc和ESP分别是吉布斯自由能热校正和单点能)。S−Ala_1 与Mg2+的配合物S−Ala_1·Mg2+记作S−A_1;S−A_1 在a 通道异构的第1 个S−型过渡态配合物记作S−T1_1a,第1 个S−型中间体配合物记作S−I1_1a,a 和b 通道共用的结构X 记做Xa(b),4 个水分子与S−A_1a(b)的Mg2+配位同时2 个水分子簇(二聚水)与S−A_1a(b)形成氢键记作S−A_1a(b)←4H2O·(H2O)2,其他体系表示法相似。计算采用Gaussian09[27]程序。
2 结果与讨论
文献[13]的气相Ala 分子最稳定的两对手性对映体及Mg2+的构象见图1,S−Ala_1 相对于S−Ala_2 的吉布斯自由能是3.0 kJ/mol[13],后者稳定,气相环境下后者的分布高于前者。
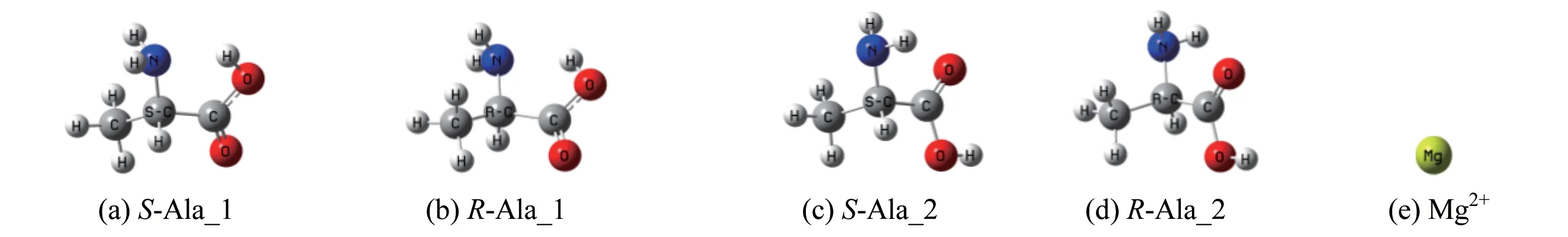
图1 Ala分子手性对映体及Mg2+的几何构型Fig.1 Chiral enantiomer of Ala and geometric configuration of Mg2+
Mg2+可在不同位置与S−Ala_1 和S−Ala_2 的氮和氧配位形成配合物,计算表明,Mg2+与S−Ala_1的羧基二配位,羧羟基氢迁移到氨基氮的螯合物S−A_1 最稳定;Mg2+与S−Ala_2 的羰基氧及氨基氮二配位的螯合物S−A_2 最稳定。优化的2 个螯合物见图2,下面对S−A_1 和S−A_2 的手性转变分别进行讨论。

图2 配合物S−A_1和S−A_2的几何构型Fig.2 Geometric configuration of S−A_1 complex and S−A_2 complex
2.1 S−A_1的手性转变
S−A_1 的手性转变可以在3 个通道a、b 和c 实现,反应历程见图3,反应过程的势能面见图4。下面分别进行讨论。
2.1.1 a 和b 通道这两个通道共用第1 基元,第1 基元反应:S−A_1 经过渡态T1_1a(b),13H 沿过渡态矢量的正向从α−碳1C 迁移到羰基氧11O,构型异构成中间体配合物I1_1a(b)。从S−A_1 到T1a(b),1C—13H键的键长从0.109 9 nm拉伸至0.153 0 nm 断裂,二面角4C—1C—6N—9C 的键角从−125.0°变为−144.6°,C—H键的大幅拉伸断裂及骨架的形变需要的能量很高,T1a(b)产生了297.5 kJ/mol 的内禀能垒。这与气相Ala 在此过程的内禀能垒290.0 kJ/mol 相差不明显,说明Mg2+的配位对此氢迁移反应影响不大。
接下来的反应历程分为a分通道和b分通道。
1) a分通道
第2 基元反应:I1_1a(b)经过渡态T2_1a,13H在纸面里沿过渡态矢量的正向从11O 迁移到α−碳,构型异构成R−型产物配合物R−A_1a。结构分析表明,R−A_1a与S−A_1 镜像对称,说明至此S−A_1实现了手性对映体转变。从I_1a(b)到T2_1a,11O—13H 键的键长从0.097 3 nm 拉伸至0.124 3 nm 断裂,O—H 拉伸幅度不是很大,又O—H 键较C—H键容易断裂,T_1a产生的内禀能垒是192.8 kJ/mol,比T_1a(b)产生的内禀能垒低许多。这与气相Ala在此基元反应的内禀能垒194.2 kJ/mol 相差无几,说明说明Mg2+的配位对此氢迁移反应基本无影响。
纵观S−A_1 在a 通道上反应历程的驻点构象和势能面可以看出,所有驻点构象及各个驻点的能量关于I1_1a(b)对称,体现了S−A_1 在该通道上实现手性对映体转变的过程美和内在美。
2) b分通道
第2基元反应:I1_1a(b)经过渡态T2_1b,8H 在纸面内侧从氨基N 向α−C 迁移,构型异构,形成R−型中间体配合物R−I2_1b,S−A_1 在b 通道实现了手性转变。从I1_1a(b)到T2_1b,6N—8H 键的键长从0.103 0 nm 拉伸至0.138 7 nm 断裂,T2_1b产生的内禀能垒是212.5 kJ/mol。该能垒与气相下单体Ala 在此基元反应的内禀能垒106.5 kJ/mol[9](在本工作理论水平下是109.6 kJ/mol)相比较显著升高,Mg2+对此基元反应表现出极强的负催化作用。原因之一是Mg2+与羧基氧10O 和11O 二配位,导致8H的NPA电荷从0.436 e显著增加到0.493 e,6N 的NPA 电荷从0.795 e 增加到0.799 e,α−碳1C的NPA 电荷从−0.245 e 骤降到−0.037 e,因此6N对8H 的库仑引力显著增加,1C 对8H 的库仑显著减小。原因之二是单体INT1的偶极矩在6N—1C方向的分量向上,质子从质子化氨基向α−碳迁移是逆着偶极矩矢量在6N—1C 方向上的分量,使体系的偶极矩减小;而I1_1a(b)的偶极矩在6N—1C 方向的分量向下,质子从质子化氨基向α−碳迁移是顺着偶极矩矢量在6N—1C 方向上的分量,使体系的偶极矩增大,前者相对容易。
从第3基元反应开始又分为b1和b2分通道。
①b1分通道
第3 基元:R−I2_1b经过渡态R−T3_1b1,9C−1C键右视顺时针内旋转,异构成配合物R−I3_1b1。从R−I2_1b1到R−T3_1b1,二面角10O—9C—1C—6N 的键角从25.9°增加到−26.5°,9C—1C 键旋转了52.4°,碳—碳键内旋转所需能量很小,R−T3_1b1产生的内禀能垒只有4.7 kJ/mol。
第4 基元反应:R−I3_1b1经过渡态R−T4_1b1,9C—1C 键右视顺时针内旋转,异构成产物配合物R−A_1b1,至此S−A_1a(b)实现了手性对映体转变。从R−I3_1b1到R−T4_1b1,二面角11O—9C—1C—6N的键角从82.5°减小增加到71.9°,9C—1C 键内旋转了10.6°,碳—碳键的小幅内旋转所需能量不多,R−T4_1b1产生的内禀能垒只有2.6 kJ/mol。结构分析表明,R−A_1b1与S−A_1 镜像对称,全同于R−A_1a,记作R−A_1a(b1)。
②b2分通道
第3 基元:R−I2_1b经9C—10O—14Mg 做剪式运动的过渡态R−T3_1b2,11O—14Mg 配位键断裂,6N—14Mg 配位键形成,异构成中间体配合物R−I3_1b2。从R−I2_1b到R−T3_1b2,较弱的配位键11O—14Mg(键长是0.218 5 nm)断裂,9C—10O—14Mg 键的键角从101.5°小幅增加到117.2°,所需能量很小,R−T3_1b2产生的内禀能垒只有7.1 kJ/mol。
第4基元反应:R−I3_1b2经过渡态R−T4m_1b2或R−T4n_1b2,11O—9C 键仰视顺时针内旋转,异构成产物配合物R−A_2_1b2, 至此在b2 分通道实现了S−A_1 的手性转变。从R−I3_1b2到R−T4m_1b2,二面角13H—11O—9C—10O 键的键角从−178.4°变为99.3°,11O—9C键的内旋转所需能量不是很多,R−T5mb2产生的内禀能垒是35.8 kJ/mol。相似的R−T5nb2产生的内禀能垒是36.2 kJ/mol。结构分析表明,R−A_2_1b2是S−A_2 的手性对映体,因此,b2分通道给出了S−A_1向S−A_2的手性对映体异构的机理。
2.1.2 c通道
第1基元反应:S−A_1经过渡态T1_1c,13H 沿过渡态矢量的正向从α−碳1C 迁移到氨基氮6N,12H 从氨基氮6N 迁移到羰基氧10O,构型异构成中间体配合物I1_1c。从S−A_1 到T1_1c,1C—13H键的键长从0.109 9 nm 拉伸至0.127 6 nm 断裂;6N—12H 断裂,12H 从6N 迁移到10O;二面角4C—1C—6N—9C 的键角从−125.0°变为−166.4°,C—H 键的拉伸断裂、N—H 断裂及骨架形变需要的能量很高,T1_1c产生了337.7 kJ/mol 内禀能垒。这与气相下单体中性的Ala_1 的α−H 从α−碳向氨基氮迁移的内禀能垒250.0 kJ/mol(文献[9]理论水平下约是267.0 kJ/mol,250.0 kJ/mol 是在本工作理论水平下计算的值)相比较显著增加,说明Mg2+对此氢迁移反应起了较强的负催化作用。原因是S−Ala_1 与Mg2+螯合后,S−Ala_1 从中性变为两性,此基元反应的反应物到过渡态从单质子迁移变成了双质子迁移,过渡态的稳定性也陡然减小。此基元反应是双质子协同非同步迁移过程,T1_1c靠近产物配合物I1_1c,是晚期过渡态。
第2 基元反应:I1_1c经过渡态T2_1c,氨基沿过渡态矢量的负向俯视顺时针旋转,12H从纸面里翻转到纸面外,构象异构成中间体配合物I2_1c。从I1_1c到T2_1c,1C—6N 键内旋转43.6°,10O—9C 键内旋转47.5°,C—N 键及O—C 键的内旋转所需能量很小,T2_1c产生的内禀能垒只有1.1 kJ/mol,此基元几乎无势垒。
第3 基元反应:I2_1c经过渡态T3_1c,8H 从氨基氮6N 迁移到α−碳1C,12H 羧基氧10O 迁移到氨基氮6N,异构成R−A_1c。从I2_1c到T3_1c是1个质子迁移,T3_1c与T1_1c相似(结构分析表明它们镜像对称),也很不稳定,T3_1c产生了185.8 kJ/mol的内禀能垒。该能垒低于T1_1c产生的能垒,主要原因是从I2_1c到T3_1c过程是单质子迁移(8H 从6N 向1C 迁移),T3_1c靠近反应物,是前期过渡态。结构分析表明,R−A_1c与S−A_1 镜像对称,全同于R−A_1a(b1),记作R−A_1a(b1,c)。
S−A_1 在c 通道上反应历程的驻点构象和势能面关于T2_1c对称。
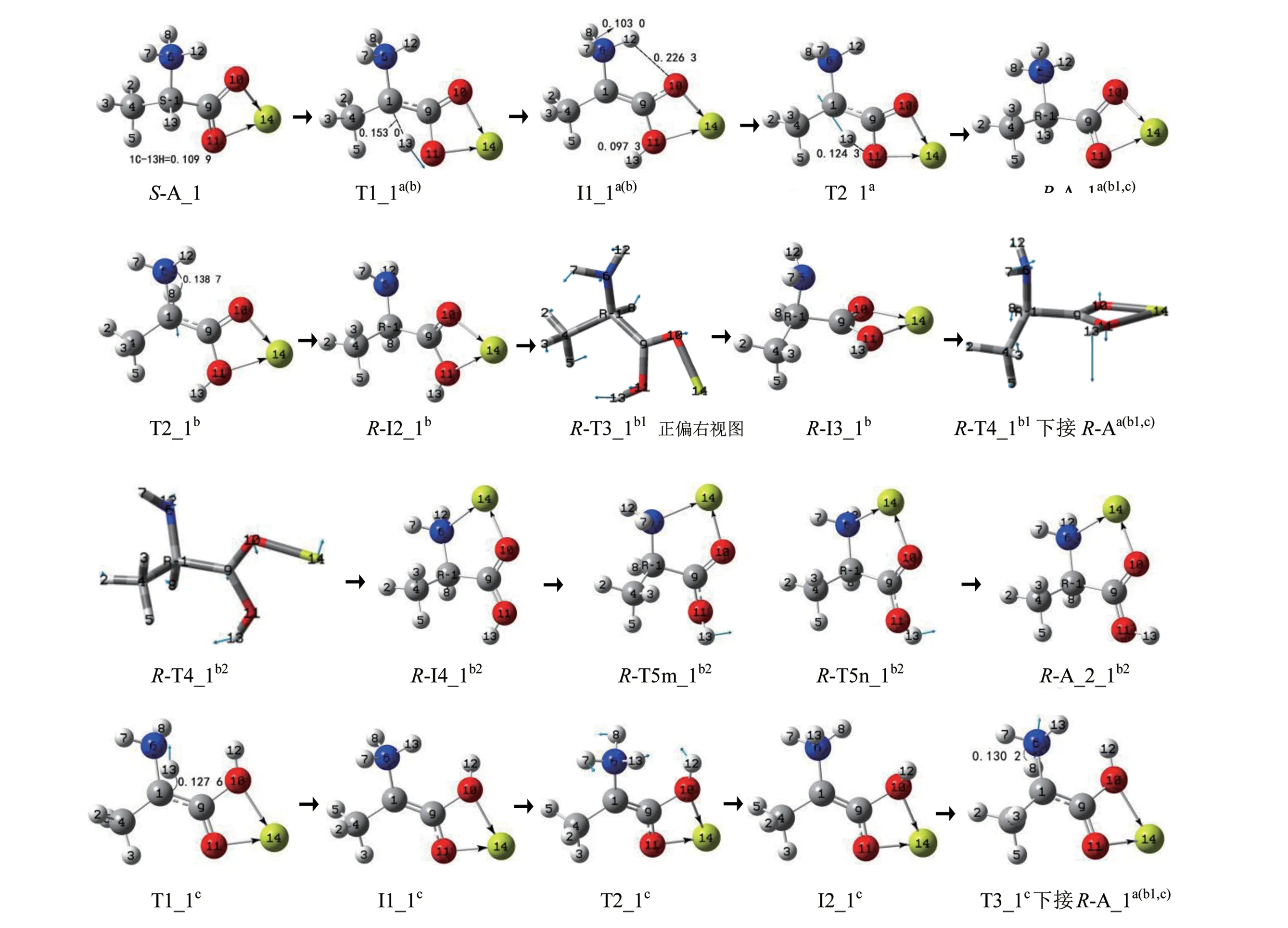
图3 S−A_1的手性转变历程(键长单位:nm)Fig. 3 Reaction process of chiral transition of S−A_1(Bond length unit:nm)

图4 S−A_1手性转变反应的吉布斯自由能势能面(括号内分子的能量是括号内的数据)Fig.4 Gibbs free energy surfaces of chiral transition reaction of S−Ala
从图4 可以看出,S−A_1 在a,b,c 通道手性转变反应的活化能(表观能垒)(气相反应的活化能取表观能垒,后面相似之处不再解释!)分别是297.5、317.2 和337.7 kJ/mol,a 通 道 具 有 优 势。S−A_1 在a 通道手性转变反应的活化能高于单体Ala 手性转变优势通道的活化能266.0 kJ/mol[13](在本文理论水平下是257.5 kJ/mol),说明Mg2+对S−Ala 的手性转变具有负催化作用,亦即Mg2+与Ala_1 配位后,可以极好地保持其旋光性(手性特征)。
从图4 及前面的讨论可知:a 是S−A_1 手性转变反应的优势通道,是α−H 以羰基O 为桥迁移。而单体Ala 手性转变的优势通道是α−H 以氨基N 为桥迁移[13],亦即Mg2+的存在改变了S−Ala手性转变反应的机理。两性配合物R−A_1a(b1,c)的稳定性好于中性的R−A_2b2,更是好于中性的Ala_1 与Mg2+的配合物,而文献[28]的研究表明气相环境下Ala的两性离子不存在,因此,Mg2+与Ala的配位显著地改变了Ala的稳定构象及稳定性的排序。
2.2 S−A_2的手性转变
S−A_2 的手性转变反应有3 个通道a、b 和c,反应历程、驻点构象及过渡态矢量见图5,反应的吉布斯自由能势能剖面,见图6,先讨论a、b 通道,而后讨论c通道。
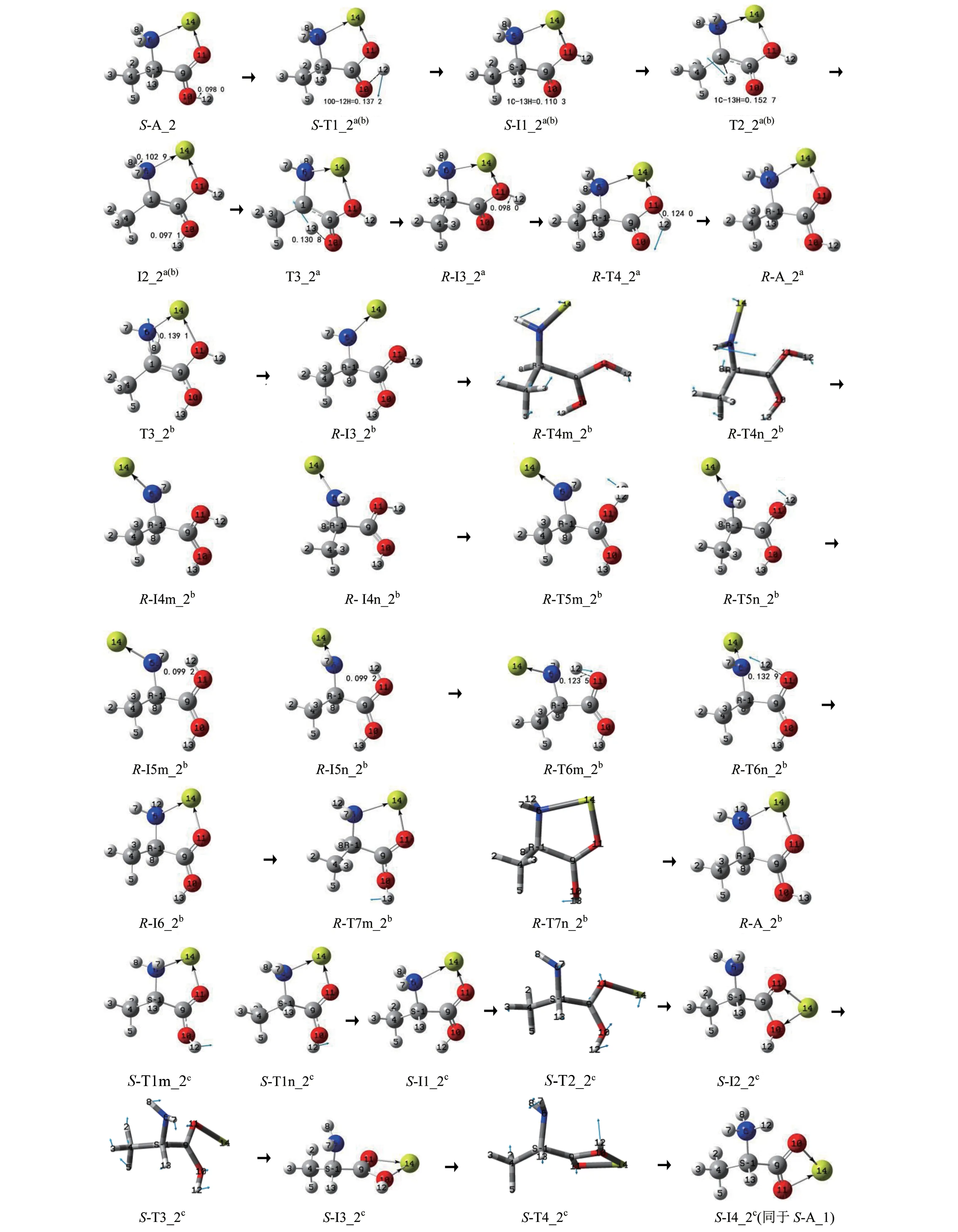
图5 S−A_2的手性转变历程(键长单位:nm)Fig.5 Reaction process of chiral transition of S−Ala_2 (Bond length unit:nm)
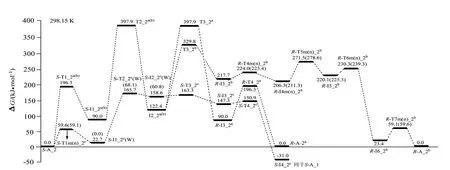
图6 S−A_2手性转变反应的吉布斯自由能势能面(括号内分子的能量是括号内的数据)Fig.6 Gibbs free energy surfaces of chiral transformation reaction of S−Ala_2
2.2.1 a和b通道共用第1和第2基元
第1 基元反应。S−A_2 经过渡态S−T1_2a(b),12H 从10O 向11O 的迁移,异构成第一中间体配合物S−I1_2a(b)。从S−A_2 到S−T1_2a(b),10O—12H键的键长从0.098 0 nm 拉伸至0.137 2 nm 断裂,10O—12H 键的断裂使S−T1_2a(b)产生了196.3 kJ/mol 的内禀能垒。这与气相Ala 在此基元反应的内禀能垒135.1 kJ/mol[5](在本工作理论水平下)相比较明显升高,说明Mg2+的存在对此基元反应具有显著的负催化作用。原因是S−A_2 的偶极矩方向向上,S−Ala_2 的偶极矩方向向下,并且前者的数值(10.173 8 D)远大于后者(1.358 7 D),导致S−A_2 的羧羟基H(带正电)向羰基氧迁移(偶极矩增大)比S−Ala_2 的羧羟基H(带正电)向羰基氧迁移(偶极矩减小)难度大许多。
第2 基元反应。S−I1_2a(b)经过渡态T2_2a(b),13H 从α−C 向羰基氧10O 迁移,异构成羧基质子化的中间体配合物I2_2a(b)。从S−I1_2a(b)到T2_2a(b),1C—13H 键的键长从0.110 3 nm 拉伸至0.152 7 nm断裂, 二 面 角4C—1C—6N—9C 的键角从−125.7°变为−142.2°,C—H 键的断裂及骨架的形变要给予很多的能量,T2_2a(b)产生了307.9 kJ/mol 的内禀能垒。这与气相Ala 在此基元反应的能垒296.3 kJ/mol[5]相比较,在误差允许的范围内可以认为没变,说明Mg2+的存在对此基元反应几乎无影响。
1) a通道
第3 基元反应。I2_2a(b)经过渡态T3_2a,实现了13H 在纸面内侧从质子化羧基的10O 向α−碳1C迁移,异构成R−型中间体配合物R−I3a_2,S−A_2实现了手性转变。从I2_2a(b)到T3_2a,10O—13H键的键长从0.097 1 nm 拉伸至0.130 8 nm 断裂,T3_2a产生的内禀能垒是275.5 kJ/mol。此能垒与文献[9]中气相S−Ala_1 手性转变在此基元的内禀能垒189.5 kJ/mol 相比明显升高,原因有2 个:一是气相下单体和Mg2+催化的此基元反应都是偶极矩增大的过程,I2_2a(b)的偶极矩(10.677 3 D)大于对应的气相的中间体INT2_1 的偶极矩(3.428 4 D)很多,I2_2a(b)比INT2_1 的13H 在纸面内侧从10O 向1C 迁移困难得多;二是I2_2a(b)与INT2_1相比较,α−C 上的负电荷从−0.114 e 降到−0.022 e,13H 与α−C 的距离从0.248 9 nm 增加到0.265 5 nm,13H 的正电荷量增加很少,导致I2_2a(b)的13H与α−C的库仑引力下降。
第4 基元反应。R−I3_2a经过渡态R−TS4_2a,实现了12H 在羧基内的回迁,异构成R−型产物配合物R−A_2a。结构分析表明,R−A_2a与S−A_2 镜像对称,R−A_2a实现了手性对映体转变。从T3_2a到R−TS4_2a,11O—12H 键的键长从0.098 0 nm 拉伸至0.124 0 nm 断裂,TS4_2a产生的能垒是106.3 kJ/mol。此基元的能垒远小于第一基元的能垒,原因有二,一是R−I3_2a的偶极矩方向向上,并且数值较大,此基元反应是H 逆着偶极矩的方向迁移;二是此基元比第一基元O—H 键的拉伸幅度小。此基元的能垒也小于单体Ala 在此基元的内禀能垒135.1 kJ/mol[5],原因与第1 基元相反,不再赘述。综上,Mg2+的存在对此基元的质子迁移反应具有较好的正催化作用。
S−A_2 在a 通道反应历程的驻点构象和势能面关于I2_2a(b)对称。
2) b通道
第3基元反应。I2_2a(b)经过渡态T3_2b,8H 在纸面里从氨基N 向α−C 迁移,异构成R−型中间体R−I3_2b,S−A_2 实现了手性转变。从I2_2a(b)到T3_2b,6N—8H 键 的 键 长 从0.102 9 nm 拉 伸 至0.169 1 nm 断裂,T3b_2 产生的内禀能垒是207.4 kJ/mol。这与气相Ala 在此基元的内禀能垒109.6 kJ/mol[12]相比大幅度升高,说明Mg2+对此基元有极强的负催化作用。原因与2.1 中b 通道第3 基元相似,不再赘述。
第4基元反应。R−I3_2b经过渡态R−TS4m_2b或R−TS4n_2b,6N—1C 键俯视顺时针或逆时针内旋转,异构成中间体配合物R−I4m_2b或R−I4n_2b。从R−I3_2b到R−TS4m_2b,二面角14Mg—6N—1C—9C 的 键 角 从4.0°变 为−50.8° (6N—1C 内 旋 转54.8°),化学键的内旋转所需能量很少,R−TS4m_2b产生了6.3 kJ/mol 的内禀能垒。相似的R−TS4n_2b产生的内禀能垒是5.7 kJ/mol。
第5 基元反应。R−I4m_2b经过渡态R−T5m_2b,羧羟基旋转(11O—9C 内旋转),异构成R−I5m_2b。从R−I4m_2b到R−T5m_2b,O=C 双键内旋转,二面角12H—11O—9C—10O 键的键长从0.3°变为97.3°,同时羧羟基H 向羧基内旋转还要克服来自于Mg2+的少许空间位阻,所需能量不高但也不是很低,R−T5m_2b产生了65.2 kJ/mol 的内禀能垒。相似的R−I4n_2b经过渡态R−T5n_2b,构象异构成R−I5n_2b。R−T5n_2b,产 生 的 内 禀 能 垒 是67.3 kJ/mol。
第6 基元反应。R−I5m_2b经过渡态R−T6m_2b,12H 沿过渡态矢量的负方向从11O 迁移到6N,异构 成R−I6_2b。从R−I5m_2b到R−T6m_2b,11O—12H 键的键长从0.099 2 nm 拉伸至0.123 5 nm 断裂,此过程R−T6m_2b产生的内禀能垒很小,只有10.2 kJ/mol。相似的R−I5n_2b经过渡态R−T6n_2b,异构成R−I6_2b,R−T6n_2b产生的内禀能垒是14.0 kJ/mol。
第7基元反应。R−I6_2b经过渡态R−TS7m_2b或R−TS7n_2b,10O—9C 键仰视顺时针或逆时针内旋转,异构成产物配合物R−A_2b。此基元的羧羟基旋转相似于第5 基元,但没有空间位阻,比较而言,R−TS7m_2b和R−TS7n_2b产生的内禀能垒小很多,分别是35.7和36.2 kJ/mol。
2.2.2 c通道
第1 基元反应。S−A_2 经过渡态S−T1m_2c或S−T1n_2c,10O—9C 内旋转,12H 从羧基内侧转到外侧,异构成中间体S−I1_2c。此基元正反应相似于b通道第7基元的逆反应,结构分析表明,S−I1_2c与R−I6_2b镜像对称,不再赘述。S−T1m_2c或S−T1n_2c产生的内禀能垒分别是59.6和59.1 kJ/mol。
第2 基 元 反 应。S−I1_2c经 过 渡 态S−T2_2c,14Mg—6N 配位键断裂与14Mg—119C 沿虚频正向剪式运动协同进行,异构成S−I2_2c。从S−I1_2c到S−T2_2c,强配位键14Mg—6N 断裂、14Mg—11O—9C 键翻转124.6°及14Mg 从羧基的外侧摆动到内侧(S−I1_2c的5 元环结构破坏),需很多能量,S−T2_2c产生了143.0 kJ/mol的内禀能垒。
第3 基 元 反 应。S−I2_2c经 过 渡 态S−T3_2c,9C−1C 键右视顺时针内旋转,异构成中间体S−I3_2c。结构分析表明,此基元与2.1节中b1通道的第4 基元镜像对称,S−T3_2c产生了4.7 kJ/mol 的内禀能垒。
第4 基元反应。S−I3_2c经过渡态S−T4_2c,异构成两性配合物S−I4_2c。结构分析表明,S−I4_2c全同于S−A_1a(b1,c)。结构分析表明,此基元反应与2.1 节中b1 通道的第5 基元相似,S−T4_2c产生的内禀能垒是3.6 kJ/mol,不再赘述。S−I4_2c接下来的异构亦即S−A_1a(b1,c)的异构,见2.1节。
从图6 可以看出,S−A_2 在a 和b 通道上手性转变的表观能垒是397.9 kJ/mol,图6 结合图4 可以看出,S−A_2 在c 通道上S−I4_2c接S−A_1a(b)的手性转变的表观能垒是297.5−31.0=266.5 kJ/mol。反应通道c 具有优势,266.5 kJ/mol 高于单体Ala_2手性转变优势通道的表观能垒249.3 kJ/mol(249.3 kJ/mol 是在本工作理论水平下计算的值,在文献[8]理论水平下是266.0 kJ/mol),说明Mg2+对S−Ala_2 的手性转变具有负催化作用。266.5 kJ/mol 的能垒很高,通常难以逾越,说明S−Ala_2与Mg2+配位后可以很好地保持其手性特征。
2.3 水分子(簇)作用下S−A_1 和S−A_2 手性转变反应决速步及水溶剂效应
从前面的讨论可知,S−A_1和S−A_2手性转变反应优势通道的表观能垒(活化能)均来自于过渡态T1_1a(b),S−A_1手性转变反应优势通道的第2基元与第一基元对称,因此,为节省篇幅,只讨论水分子(簇)作用下的S−A_1→T1_1a(b)→T1_1a(b)基元反应及溶剂效应。
水分子与S−A_1 的作用有两种,一是水分子(簇)会与Mg2+形成配位键,二是水分子(簇)会与S−A_1 的氢及氧形成氢键。由于配位键强于氢键,因此实际的情况应是水分子(簇)与Mg2+配位饱和后,再与S−A_1 的氢及氧形成氢键。计算表明,Mg2+与O 最多可以6 配位,水分子(簇)与S−A_1 的Mg2+最基本的配位形式可以有2 种,一是Mg2+与羧基氧二配位后再与4 个水分子配位,二是Mg2+与羧基氧一配位后再与5 个水分子配位(其中有1个水分子与Mg2+配位的同时还与羧基氧氢键作用)。水分子(簇)与Mg2+配位饱和后,可以有1个、2 个或3 个水分子簇与S−A_1 的氢及氧形成氢键,作为质子12H 从1C 向10O 迁移的媒介。计算表明,本基元反应3个水分子簇作H 迁移媒介并不比2 个水分子簇作媒介具有优势。因此为节省篇幅,对于第1 种情形只讨论1 个和2 个水分子簇与S−A_1 的氢及氧形成氢键,作为质子12H 从1C 向10O 迁移媒介的情况,反应物记作S−A_1←4H2O·H2O和S−A_1←4H2O·(H2O)2;对于第2种情形只讨论2 个水分子簇与S−A_1 的氢及氧形成氢键,作为质子12H 从1C 向10O 迁移媒介的情况,反应物记作S−A_1←5H2O·(H2O)2。另外,为充分考虑水分子簇的作用,对第2 种情况,增加了1 个水分子与羰基氧和与Mg2+配位的1 个水分子同时氢键作用的讨论,反应物记作S−A_1←5H2O·H2O·(H2O)2。上述4个反应物经相应的过渡态实现质子转移的反应历程见图7,反应过程的势能面见图8,下面分别经行讨论。
2.3.1 S−A_1←4H2O·H2O 经S−T1_1a(b)←4H2O·H2O 到T1_1a(b)←4H2O·H2O 过程从S−A_1←4H2O·H2O 到S−T1_1a(b)←4H2O·H2O,键1C—12H 的键长从0.109 9 nm 拉伸至0.175 7 nm 断裂,键27O—28H 的键长从0.096 4 nm 拉伸至0.107 1 nm 断裂,二面角4C—1C—6N—9C 的键角从−125.7°变为−136.6°,C—H 和O—H 的键断及骨架形变需很多能量,但结构分析表明,过渡态S−T1_1a(b)←4H2O·H2O 六元环存在两个较强的氢键,且六元环结构共面程度较好,增加了过渡态的稳定性,因此S−T1_1a(b)←4H2O·H2O 产生了195.6 kJ/mol 的内禀能垒,这与2.1节中讨论的没有水分子作用时此基元反应的能垒297.5 kJ/mol 相比较大幅减小,说明水分子起了很好的催化作用。溶剂化计算表明,水溶剂环境下的能垒是192.9 kJ/mol,水溶剂效应影响可以忽略。
2.3.2 S−A_1←4H2O·(H2O)2经S−T1_1a(b)←4H2O·(H2O)2到T1_1a(b)←4H2O·(H2O)2过程从S−A_1←4H2O·(H2O)2到S−T1_1a(b)←4H2O·(H2O)2,键1C—12H的键长从0.109 6 nm拉伸至0.182 3 nm断裂,键14O—19H 的键长从0.097 5 nm 拉伸至0.123 3 nm 断裂,键17O—16H 的键长从0.097 3 nm 拉伸至0.1079 nm 断裂,骨架二面角4C—1C—6N—9C 的键角从−120.9° 变为−140.3°,C—H 和O—H 的断裂及骨架的形变需一定的能量,但结构分析表明,八元环过渡态S−T1_1a(b)←4H2O·(H2O)2的三个氢键很强,且过渡态的八元环结构共面程度很好,过渡态的稳定性很好,因此S−T1_1a(b)←4H2O·(H2O)2产生了172.3 kJ/mol 的内禀能垒,与没有水分子作用时此基元反应的能垒297.5 kJ/mol 相比较显著减小,说明2 个水分子簇起了很好的催化作用。溶剂化计算表明,水溶剂环境下的能垒是172.5 kJ/mol,水溶剂效应影响可以忽略。
2.3.3 S−A_1←5H2O·(H2O)2经S−T1_1a(b)←5H2O·(H2O)2到T1_1a(b)←5H2O·(H2O)2过程从S−A_1←5H2O·(H2O)2到S−T1_1a(b)←5H2O·(H2O)2,键1C—12H的键长从0.109 9 nm拉伸至0.169 7 nm断裂,键14O—19H 的键长从0.097 6 nm 拉伸至0.119 5 nm 断裂, 但 键17O—16H 的键长从0.097 1 nm 拉伸至0.110 6 nm 断裂,骨架二面角4C—1C—6N—9C 从−126.3°变为−137.7°,C—H 和O—H 的断裂拉伸及骨架的形变幅度小于从S−A_1←4H2O·(H2O)2到S−T1_1a(b)←4H2O·(H2O)2过程,八元环过渡态S−T1_1a(b)←5H2O·(H2O)2的三个氢键也很强,八元环结构共面程度也很好,过渡态的稳定性也很好,因此S−T1_1a(b)←4H2O·(H2O)2产生了157.9 kJ/mol 的内禀能垒,小于S−T1_1a(b)←4H2O·(H2O)2产生的能垒。溶剂化计算表明,水溶剂环境下的能垒是157.5 kJ/mol,水溶剂效应影响可忽略。
2.3.4 S−A_1←5H2O·H2O·(H2O)2 经S−T1_1a(b)←5H2O·H2O·(H2O)2到T1_1a(b)←5H2O·H2O·(H2O)2过程结构分析表明,从S−A_1←5H2O·(H2O)2到S−T1_1a(b)←5H2O·(H2O)2,键1C—12H 和14O—19H 的键长拉伸基本同于前一个过程,但键17O—16H 的拉伸大于前一个过程,骨架二面角4C—1C—6N—9C 的形变大于前一过程,因此S−T1_1a(b)←5H2O·H2O·(H2O)2产生了189.1 kJ/mol 的内禀能垒,大于S−T1_1a(b)←5H2O·(H2O)2产生的能垒。溶剂化计算表明,水溶剂环境下的能垒是182.8 kJ/mol,水溶剂效应影响也可忽略。

图7 水分子(簇)作用下S−A_1→T1_1a(b)→T1_1a(b)基元反应的历程Fig.7 Reaction process of S−A_1→T1_1a(b)→T1_1a(b) pathway with water molecules (clusters)
从图8可以看出,水汽相下水分子(簇)作用下S−A_1→T1_1a(b)→T1_1a(b)基元质子迁移反应的最低能垒是157.9 kJ/mol,接近反应的极限能垒160.0 kJ/mol[29],水汽相下S−A_1 只能痕量的消旋。从图8 和图6 可以看出,水汽相下S−A_2 在优势通道上手性转变的活化能(表观能垒)是165.7 kJ/mol,水汽相下S−A_2 通常不能消旋。从图8 还可以看出,水液相下水分子(簇)作用下S−A_1→T1_1a(b)→T1_1a(b)基元质子迁移反应的最低能垒是157.5 kJ/mol,接近反应的极限能垒160.0 kJ/mol[29],水液相下S−A_1 难以消旋或只能痕量的消旋。与水液相环境下两性Ala 手性转变决速步能垒130.5 kJ/mol(130.5 kJ/mol这是在本工作理论水平下的值,在文献[12] 理论水平下是141.8 kJ/mol)相比较可知,水液相环境下Mg2+对两性Ala的旋光异构反应有负催化作用。
为了确定水液相下S−A_2 在优势通道c 上手性转变的活化能,要计算出c 通道的最高内禀能垒,因此,计算了S−A_2 手性转变优势通道c 上第2 基元反应S−I1_2c→S−T2_2c→S−I2_2c过程的溶剂化势能面,见图6 的c 通道第2 基元括号内的数据。从图6 的c 通道第2 基元括号内的数据可知,水溶剂环境下S−I1_2c→S−T2_2c→S−I2_2c基元反应的内禀能垒是68.1 kJ/mol。因此,水溶剂环境下S−A_2在手性转变优势通道c上的活化能应是水液相下水分子(簇)作用下S−A_1→T1_1a(b)→T1_1a(b)基元质子迁移反应的最低能垒157.5 kJ/mol,水液相下S−A_2也只能痕量的消旋。
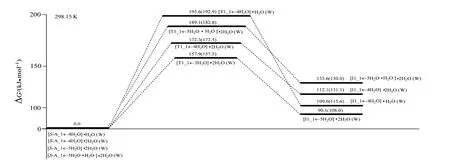
图8 水分子(簇)作用下S−A_1→T1_1a(b)→T1_1a(b)基元反应的吉布斯自由能势能面Fig.8 Gibbs free energy potential surfaces of S−A_1→T1_1a(b)→T1_1a(b) pathway with water molecules (clusters)
综合图6 和图8 可知,水溶剂环境下丙氨酸Mg2+螯合物主要以两性离子形式存在,并且其手性特征很难改变。丙氨酸Mg2+螯合物可以作为理想的补充丙氨酸及镁离子的药品或营养品。
3 结 论
在M06/6−311++G (2df, pd)//M06/6−31++G(d,p)双水平结合自洽反应场理论的SMD 模型方法对标题反应进行了研究,得到如下结论。
1)Mg2+与Ala 的2 个羰基氧二配位的两性离子螯合物A_1 最稳定,Mg2+与Ala 的氨基氮和羰基氧二配位形成的中性离子螯合物A_2的稳定性次之。
2)S−A_1 的手性转变有3 个通道a、b、c,分别是α−H 只以羰基氧为桥、以羰基氧与氨基氮联合为桥和只以氨基氮为桥迁移;S−A_2的手性转变也有3 个通道a、b、c,a 和b 分别是羧基内H 迁移后α−H 只以羰基氧为桥和羰基氧与氨基氮联合为桥,c 通道是S−A_2 经Mg2+与氨基氮的配位键断裂和1C−9C内旋转向S−A_1异构后的异构。
3)气相的S−A_1 和S−A_2 手性转变优势通道的反应活化能在266.5 kJ/mol 以上;在水分子(簇)作用下S−A_1 和S−A_2 手性转变优势通道的反应活化能在157.9 kJ/mol以上;水溶剂环境下S−丙氨酸Mg2+螯合物主要以S−A_1的构象存在,手性转变优势通道的反应活化能为157.5 kJ/mol。
结果表明,通常气相、水汽相及水液相环境下丙氨酸Mg2+都可以很好地保持其手性特征。
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
科学导报(2018年30期)2018-05-14 12:06:01
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
Chinese Journal of Chemical Engineering(2014年3期)2014-07-24 15:40:13
