
Ni原子表面吸附和笼内封装掺杂硼富勒烯B40的功能化应用研究*
2020-10-20 09:39谢安治文天珍李继玲
中山大学学报(自然科学版)(中英文) 2020年5期
谢安治,文天珍,李继玲
(中山大学材料科学与工程技术学院,广东广州510275)
因信息技术飞速发展的需要,传统的微电子器件正朝高度微型化、集成化的纳米器件方向发展。低维纳米材料在纳米电子器件、纳米光电器件、纳米分子器件和高密度非挥发性存储器件等领域具有巨大的应用前景。因此,发展新型低维纳米材料成为纳米材料学和纳米电子学领域最新兴和前沿的研究热点。人们通过各种理论和实验探索合成了更多、更具优越性能的新型功能材料与纳米器件。受碳纳米材料重要应用的启发,关于非碳纳米材料性能的研究和新型非碳纳米结构的探索一直是研究的热点领域。因硼在元素周期表中与碳“最近邻”,对硼纳米管和零维硼团簇的研究得到了尤为广泛的关注,科学家试图得到类似于碳家族的更具优越性能的丰富的低维硼家族纳米材料,并在最近取得了很大的突破[1−2]。硼富勒烯和低维硼纳米结构与相应碳富勒烯和低维碳纳米材料在性能上具有很强的互补性,两者结合或许能为解决人类面临的能源和环境危机提供新的思路和技术,在储氢储锂、半导体光电应用、超导、绿色催化等领域具有重要的应用前景。人们期待能够合成类似于碳富勒烯(如C60)的硼族富勒烯笼状结构,但很长一段时间以来一直未能在实验上确定笼状硼的存在。在所有的硼富勒烯的探索中,Szwacki等[1]提出的硼富勒烯B80具有很高的对称性以及和足球C60极为相近的结构特点。然而,随后的研究表明,相对于其他幻数为80 的“核−壳”团簇异构体,足球空心笼状B80富勒烯具有相对高的能量,因而处于非稳定状态[3−4]。令人振奋的是,在最近的报导中,一种新型的全硼富勒烯笼状结构B40成功实验合成[5]。该全硼富勒烯笼状结构的发现为开发硼的新材料提供了重要线索。进一步的实验和理论研究证实了该B40全硼富勒烯团簇呈现空心笼状结构,具有典型的D2d点群对称性,其表面包含交织的双链结构、两个由6个B 原子组成的六元环和四个由7 个B 原子组成的七元环,整体恰似传统的“中国红灯笼”[6]。B40是继C60之后第二个从实验和理论上完全确认的无机非金属笼状团簇,从而成为全硼富勒烯实验和理论研究的开端。值得注意的是,硼富勒烯B40特殊的表面六元环、七元环和笼状结构特点对于外来原子或者小分子的进入和内嵌封装都非常有利。同时,较大的笼状表面也有利于外来原子在富勒烯表面的吸附。此外,在所有可作为外来原子对富勒烯进行表面修饰和内嵌封装的掺杂元素中,过渡金属元素因其常常伴随出现在低维材料的合成中,较大的共价半径和较为弥散的价电子分布等因素,使其成为外来掺杂物质的重要代表性元素。许多学者通过实验和理论工作开展了过渡金属元素的碳富勒烯掺杂应用研究,并预言了一系列良好的超导体、铁磁体、铁电体等[7−9]。鉴于以上分析,开展过渡金属元素与新型硼富勒烯B40掺杂的功能化应用研究非常重要和及时。我们希望能够通过相应的理论研究和探索,对这类材料的大规模合成和最终走向应用提供重要的理论指导。
本文通过第一原理计算系统研究了过渡金属元素Ni 对全硼富勒烯B40的外表面化学吸附及笼内内嵌封装的功能化应用。首先,根据文献中提出的硼富勒烯B40的原子排布方式,构造了B40富勒烯的笼状结构。然后,采用Ni 原子对该全硼富勒烯结构分别进行外表面化学吸附和笼内内嵌封装,并进一步计算了Ni 掺杂后的金属富勒烯结构、稳定性、电子特性和磁学性质。计算结果表明,优化后的Ni 掺杂B40金属富勒烯复合结构的平衡构型整体保持完好,吸附Ni 原子与B40表面的B 原子结合形成Ni−B 键,吸附位附近区域B−B 键的键长被拉长。Ni 原子在B40外表面吸附和笼内封装均具有较大的结合能(5.18 ~7.12 eV/atom)。从能量观点来看,Ni 原子外表面化学吸附的金属富勒烯的稳定性高于Ni 原子内嵌封装的金属富勒烯的稳定性。对金属富勒烯复合产物的电子特性和磁性计算表明,对于所有的Ni 原子掺杂的金属富勒烯复合结构,其能隙相对于全硼富勒烯B40,均有一定程度的减小;同时,磁性元素Ni 原子与全硼富勒烯作用后,磁性原子Ni 掺杂的金属富勒烯复合结构的总磁矩均为零。这种可调磁性并能同时调整材料电子能隙的性能特点,对于此类金属富勒烯在半导体分子器件方面的应用提供了理论支持和依据,具有重要的学术价值。
1 计算方法
本文采用基于密度泛函理论的第一原理计算软 件 包SIESTA 进 行 计 算[10−12]。Siesta 使 用 标 准 的Kohn−Sham 自洽密度泛函方法,结合局域密度近似(LDA−LSD)或广义梯度近似(GGA)。计算使用完全非局域形式(Kleinman−Bylander)的标准守恒赝势[13−14]。基组是数值原子轨道的线性组合(LCAO)。它允许任意个角动量、多个zeta 极化和截断轨道。计算中把电子波函数和密度投影到实空间网格中,以计算Hartree 和XC 势矩阵元素。除了标准的Rayleigh−Ritz本征态方法以外,程序还允许使用占据轨道的局域化线性组合。本次计算中,我们采用Perdew−Berke−Ernzerhof 的广义梯度近似(GGA)来描述交换关联能[15]。实空间网格划分的等效平面波截断能设定为120 Ry。自洽计算时,收敛标准设为每个原子上的力小于0.2 eV/nm。这些计算方法和理论参数的选择基于之前的相关理论计算工作,因而具有一定的合理性[16]。计算中B 元素和Ni 元素的价电子组态分别为2s2p1和3d84s2,对于B 原子和Ni 原子,分别采用double−ξ轨道和double−ξ 极化轨道展开价电子波函数。同时,对于整个笼状分子结构,建立周期性晶胞并在三个方向上均取150 nm 的真空层以避免分子之间的相互作用。整体计算过程分为两个步骤:首先,对所构造的几何结构进行充分的结构优化,得到稳定的平衡结构;然后,在稳定结构的基础上,对优化好的平衡构型开展其电子结构和自旋极化磁学性质的计算。
2 计算结果与讨论
2.1 几何结构与稳定性
首先,根据文献中对新型硼富勒烯B40结构特点和对称性的讨论[5−6],我们构造了确定的B40原子排布方式并对其构造的初始几何结构进行了充分的结构优化。优化后的B40平衡构型如图1 所示。其中,图1(a)为B40平衡构型侧视图,图1(b)为B40平衡构型俯视图。对平衡结构的分析表明,优化后的全硼富勒烯B40具有典型的中空笼状结构特点和D2d点群对称性,表面由多个相互交织的B原子双链、两个由6 个B 原子构成的六元环和四个由7 个B 原子构成的七元环所组成。如果以两个六元环作为上下底面,其侧面由四个七元环和多个交织的B 原子双链组成(Interwoven Double Chains, IDC),具有典型的“灯笼”状特征,整体结构特点与表面原子排布方式与文献中一致[5−6]。根据我们的计算,B40笼状结构的直径为0.636 nm,稍大于文献中B40笼状结构的直径0.620 nm[5]。通过对B40表面原子结合方式的分析,其表面B−B 键分为三类:六元环内B−B 键、七元环内B−B键和交织的B原子双链内的B−B键,对应三类B−B 键长的平均值分别为0.168 nm、0.164 nm 和0.171 nm。综上,通过对结构的分析,我们构造的硼富勒烯B40和优化后的平衡构型及结构参数与文献中基本一致[5−6],说明我们构造的几何构型及计算所采用的理论标准是正确和合理的。B40富勒烯特殊的表面B原子排布方式及中空笼状结构特点,有利于外来原子的表面吸附或外来原子或者小分子的笼内内嵌封装。因而,在接下来的讨论中,我们采用过渡金属磁性元素Ni 对优化后的B40进行掺杂,系统地开展了单个Ni原子在B40外表面的吸附和笼内内嵌封装的功能化应用研究。
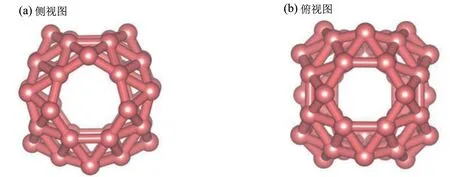
图1 B40优化平衡结构图Fig.1 The side view and top view of the optimized configuration of the pristine B40
我们选取了多个不同的对称位点作为Ni 原子的外表面吸附位和内部封装位,并对这些初始结构进行充分的几何结构优化和能量弛豫。计算结果表明,单个Ni 原子能够稳定地停留在B40的不同位置并进而形成相应的Ni 掺杂B40金属富勒烯。根据掺杂方式(外表面化学吸附和笼内内嵌封装)的不同,对应形成的Ni掺杂金属富勒烯分为两类:(1)单个Ni 原子吸附于B40的外表面所形成的金属富勒烯产物,按照传统金属富勒烯的标记方式记为“Ni−B40”金属富勒烯;(2)单个Ni原子笼内封装于B40的内嵌金属富勒烯产物,记为“Ni@B40”金属富勒烯。另外,考虑吸附位置硼原子的排布情况,为了区别同一类金属富勒烯吸附位不同,我们作了更进一步的标记,比如“Ni−B40−6in”,表示Ni 原子在B40富勒烯的表面外侧吸附且吸附位于表面B原子六元环中心内侧。
计算结果表明,对于Ni 原子笼内封装的内嵌金属富勒烯,Ni 原子有三种稳定的内嵌位置点,分别是B40表面六元环中心内侧、七元环中心内侧以及B原子交织双链交叠处中心内部。对应Ni@B40金属富勒烯复合产物分别命名为:Ni@B40−6in、Ni@B40−7in 和Ni@B40−IDC 金属富勒烯。对于Ni 原子外表面吸附于B40所形成的金属富勒烯,Ni 原子有两种稳定的吸附位置点,分别是B40表面六元环中心外侧、七元环中心外侧,对应的Ni−B40金属富勒 烯 复 合 产 物 分 别 命 名 为Ni−B40−6out 和Ni−B40−7out。优化后的平衡构型如图2(a)−(e)所示。其中,对于同一金属富勒烯构型,左边为侧视图,右边为俯视图。
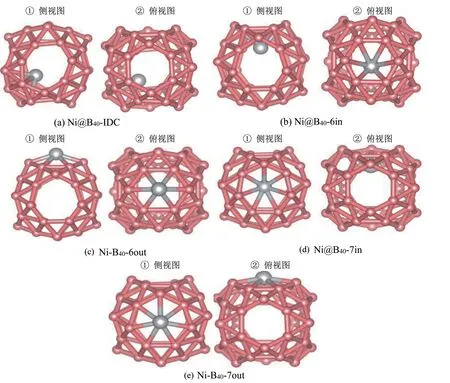
图2 结构优化后的Ni掺杂的B40金属富勒烯复合产物的平衡结构图(粉红色代表B原子,灰色代表Ni原子)Fig.2 The top view and side view of the optimized configurations of the five stable Ni−adsorbed and encapsulated B40 metallofuller⁃enes(the pink for B and the gray for Ni)
由图2可以看出,对于不同的金属富勒烯复合结构,Ni 原子吸附在不同的平衡位点并与B40表面的B 原子形成B−Ni 键。对于Ni 原子外部吸附于B40的Ni−B40−6out 金属富勒烯,Ni 原子与六环内硼原子所形成的Ni−B 键平均键长为0.201 nm,小于Ni原子和B80表面六元环B 原子所形成的的Ni−B 键平均键长0.219 nm[16]。同时,由于Ni−B 键的形成,B40表面六环内B−B 键长被拉伸,平均键长从原来的0.168 nm伸长到0.179 nm,伸长量为6.54%。
其他四种Ni掺杂的金属富勒烯结构参数如表1所示。从表1中可以看出,对于Ni原子外部吸附和笼内内嵌封装的全部金属富勒烯,Ni 与吸附点附近的硼原子均能形成稳定的Ni−B 键,且在吸附位点附近B−B 键键长引起了一定程度的伸长。因而,在B40表面发生了明显的局部结构变形。为了讨论这些结构的稳定性,我们计算了这些Ni 掺杂的金属富勒烯复合结构的结合能,结合能Eb定义如下:

其中,EB40指硼富勒烯B40的总能,ENi指单个Ni 原子的总能,ENi−B40指Ni 原子化学吸附或笼内封装的Ni 掺杂金属富勒烯的总能。每种结构的结合能如表1 所示。这里,正的Eb值表明Ni 原子对B40的外表面化学吸附和笼内内嵌封装过程均属于放热反应。从能量观点来看,对于所讨论的五种Ni 原子化学吸附和笼内封装B40的金属富勒烯复合结构,当Ni 原子化学吸附于B40外表面且位于表面七元环中心外部时,对应的Ni−B40−7out 金属富勒烯结合能为7.12 eV/atom, 因而结构最为稳定。其他四种金属富勒烯复合结构的稳定性依次是:Ni 原子吸附于B40外表面且位于六元环中心外的Ni−B40−6out 金属富勒烯,Ni 原子位于笼内交织双硼链(IDC)内侧的Ni@B40−IDC 金属富勒烯,Ni 原子位于笼内且在七元环中心内部的Ni@B40−7in金属富勒烯,最后是Ni 原子位于笼内且在六元环中心内部的Ni@B40−6in的金属富勒烯。

表1 Ni 原子外表面吸附和笼内内嵌封装B40复合结构的结构参数和结合能Table 1 The structural parameters and binding energies of the Ni adsorbed and encapsulated B40 metallofullerenes.
值得注意的是,所有Ni 原子外表面化学吸附和笼内封装的金属富勒烯复合结构的结合能都很大,远大于Ni原子与B80相互作用的结合能约3 ~4 eV[16]。因而,我们认为单个Ni 原子能够化学掺杂于全硼富勒烯B40并与之形成稳定的外表面化学吸附的Ni−B40和内部封装的Ni@B40金属富勒烯复合结构。Ni 原子的引入所引起的局部结构变形以及Ni原子与B原子之间的杂化相互作用也必将对金属富勒烯复合结构的电子特性产生大的影响。此外,Ni为重要的磁性元素。所以,在接下来的讨论中,我们重点讨论Ni 原子化学吸附和内部封装金属富勒烯复合产物的电子结构和磁学性质。
2.2 电子结构
我们计算了上述五种Ni 掺杂B40的金属富勒烯复合结构的电子结构。定义金属富勒烯笼状结构电子能隙∆Egap为最低未占据分子轨道(lowest un⁃occupied molecular orbital) 与最高占据分子轨道(highest occupied molecular orbital)之差,具体表达式为:
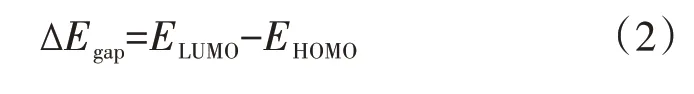
其中,EHOMO、ELUMO分别为最高占据分子轨道和最低未占据分子轨道的能级,计算结果如表2 所示。可以看出,无论Ni 原子对硼富勒烯B40的表面化学吸附还是笼内内嵌封装均可一定程度改变和调整硼富勒烯B40的能隙,并使能隙有明显减小的趋势。其中,B40@Ni−6in 金属富勒烯能隙最小,其值为0.97 eV,几乎是硼富勒烯B40能隙1.72 eV 的一半。需要指出的是,基于密度泛函理论的电子特性计算经常在一定程度上低估了材料的能隙。但,对Ni 掺杂的金属富勒烯能隙的讨论是与硼富勒烯B40的能隙相对而言的,其能隙相对差值具有同样的参考意义。因此,根据我们的计算结果,在实际材料的应用中,可以通过改变Ni 原子的掺杂位置来实现对材料电子特性调整和控制的目的。
为了进一步分析Ni 掺杂的金属富勒烯复合结构能隙变化的原因,我们计算了B40和Ni 原子掺杂后的金属富勒烯费米能级附近最高占据分子轨道(HOMO)和最低未占据分子轨道的能级(LUMO)的原子和量子轨道。图3(a)和图3(b)−(f)给出了B40和Ni 原子掺杂后的金属富勒烯分别投影到B 原子和Ni 原子上的投影态密度。其中,黑线和红线分别代表B 原子和Ni 原子的自旋极化投影态密度。竖直虚线表示硼富勒烯B40的HOMO 能级和LUMO能级,箭头所指对应每种富勒烯结构的费米能级且均已归0。这里,投影态密度采用的高斯展宽为0.02 eV。从图中可以看到,Ni 原子掺杂使得B40的HOMO 能级和LUMO 能级之间引入了新的电子态,这些电子态不仅来自于外来掺杂原子Ni,同时来自于富勒烯表面内的B原子。通过对轨道电子布局分析可知,外来Ni 原子的掺杂将使得电子从Ni 原子转移至B 原子,造成电荷的重新分布,因而使Ni 原子和B 原子间形成Ni−B 键并最终改变了Ni 掺杂金属富勒烯的局部构型。从而,金属富勒烯复合结构的电子特性得到了相应的调整,体系能隙变小。这种可通过调整外来原子吸附位置来调整金属富勒烯能隙的电子特性特点对于该类金属富勒烯作为电子材料和光电材料的应用具有重要的指导意义。
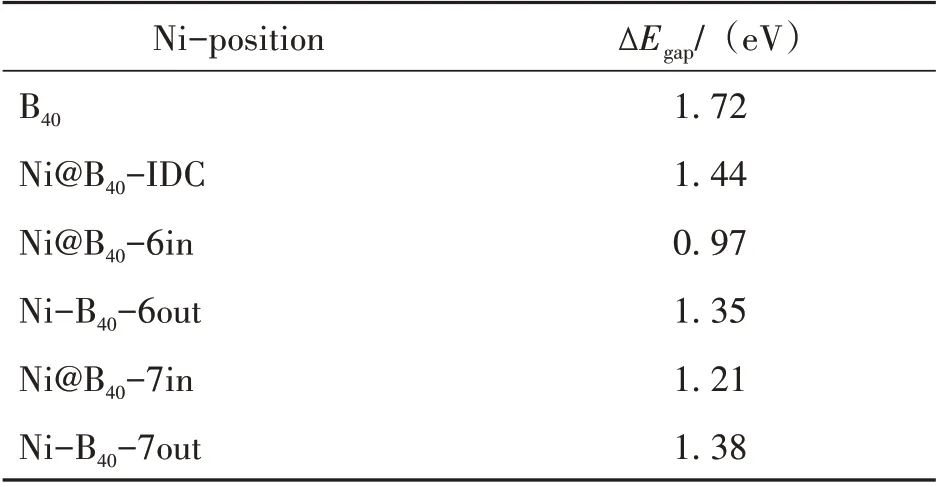
表2 全硼富勒烯B40和Ni原子外部吸附和笼内封装的电子能隙∆EgapTable 2 The energy gaps(∆Egap)of the B40 and the endohe⁃dral Ni@B40 and exohedral Ni−B40 metallofullerenes.

图3 B40和Ni原子外表面吸附和笼内封装的金属富勒烯的自旋极化投影态密度Fig.3 The density of states(DOS)and spin polarized projected density of states(PDOS)of the B40 and the Ni−doped metallofullerenes under study
2.3 磁学性质
Ni 是典型的磁性元素。在接下来的讨论中,我们通过自旋极化计算开展了对Ni 原子掺杂B40的外表面吸附和笼内内嵌封装金属富勒烯复合产物Ni−B40和Ni@B40的磁学性质讨论。所得单个Ni原子和金属富勒烯复合结构的总磁矩,如表3所示。表3中还列出了单个Ni原子和掺杂金属富勒烯后的Ni原子4s,3d,4p 轨道的电子占据情况,包括自旋向上和自旋向下。从表3可以看出,相对单个Ni原子2.00 μB的总磁矩,Ni 原子作为外部原子与富勒烯B40发生作用后,掺杂Ni 原子的磁矩变为零,发生了根本的变化和调整。同时,Ni 原子掺杂的金属富勒烯复合结构,体系总磁矩也均为零。为了更好地理解掺杂Ni 原子磁矩的变化和整个金属硼富勒烯磁学性质的情况,我们分析了金属富勒烯结构中掺杂Ni原子4s,3d,4p轨道自旋向上和自旋向下的电子占据情况,发现Ni 原子磁矩发生根本性调整的原因来自于掺杂Ni 原子的4s 轨道的自旋向上和自旋向下、3d 轨道的自旋向上电子由于自限效应而导致的占据电子的减少。表3中,随着限制效应的增强,掺杂Ni 原子4s 轨道自旋向上和自旋向下、3d 轨道自旋向上的电子向掺杂Ni 原子4p 和3d 自旋向下轨道发生明显的转移,最终导致了金属富勒烯复合结构总磁矩的根本性调整。这些计算结果和过渡磁性元素与B−N 富勒烯笼状结构B36N36,过渡元素与碳富勒烯笼以及Ni 与硼富勒烯B80所形成的金属富勒烯复合结构的磁学性质都不相同[16−19]。因而,Ni 原子掺杂的金属富勒烯复合产物Ni−B40和Ni@B40因其独特的可调电子特性和磁学性质,可作为单分子电子器件和单分子自旋电子器件,具有潜在重要应用价值。
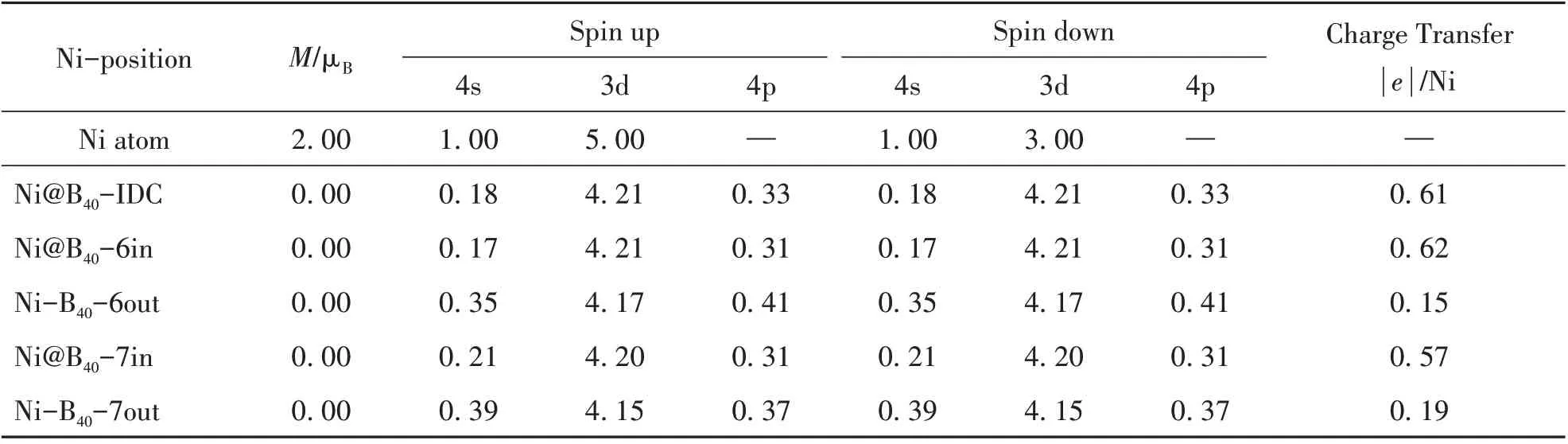
表3 金属富勒烯复合结构的总磁矩,4s、3d、4p轨道布局和Ni原子与B原子间的电荷转移Table 3 The total moments M、Mulliken population analysis of 4s,3d,4p orbitals and the charge transfer from Ni atom to B40 in the endohedral Ni@B40 and exohedral Ni−B40 metallofullerenes
3 结论
本文开展了过渡金属元素Ni 对全硼富勒烯B40的外表面化学吸附及笼内内嵌封装的功能化应用研究。计算结果表明,优化后的外来掺杂Ni 原子易于与B40表面的B 原子结合形成Ni−B 键;同时,吸附位的B−B 键的键长被拉伸从而引起了明显的局部变形。Ni 原子在B40外表面吸附和笼内封装均具有很高的结合能(5.18 ~7.12 eV/atom),说明Ni 原子易于与B40发生表面的化学吸附和笼内内嵌封装。能量计算表明,Ni 原子外表面化学吸附的金属富勒烯的稳定性高于Ni 原子内嵌封装的金属富勒烯的稳定性。对金属富勒烯复合产物的电子特性和磁性计算表明,对于所有的Ni 原子掺杂的金属富勒烯复合结构,其能隙相对于全硼富勒烯B40,均有一定程度的减小;磁性元素Ni 原子与全硼富勒烯作用后,金属富勒烯复合产物的总磁矩均为零。这种通过外来原子掺杂硼富勒烯B40可调整材料电子能隙和磁学性质的特性,对于此类金属富勒烯在半导体分子器件方面的应用提供了重要思路和理论指导。
猜你喜欢
都市人(2022年3期)2022-04-27
青岛科技大学学报(自然科学版)(2022年1期)2022-01-20
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
国际太空(2021年8期)2021-11-05
环球时报(2019-12-05)2019-12-05
中国化妆品(2019年4期)2019-11-20
太空探索(2014年4期)2014-07-19
百科知识(2009年20期)2009-11-27
