气相氟化合成氢氟烯烃催化剂的研究进展
2020-10-16 05:42张荣慧王来来谢文健
高等学校化学学报 2020年10期
张荣慧,闵 灯,王来来,谢文健
(1.中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州 730000;2.中国科学院大学,北京 100049;3.江苏理文化工有限公司,常熟 215536)
氢氟烯烃(HFOs)是有机氟化工中的重要原料和化工中间体.由于其有机氟化物的独特性质可以应用在很多方面,如化学灭火剂[1]、制冷剂[2~4]、涂料[5]及塑料制品[6,7]等.1985年,Farman等[8]发现南极上空出现臭氧空洞,其形成原因就是氟氯烃作为制冷剂、喷雾剂、发泡剂等化工制剂的大量使用.为了避免工业产品中的氟氯碳化合物对地球臭氧层持续破坏,联合国于1987年9月16日邀请所属26个会员国在加拿大蒙特利尔签署环境保护公约即《蒙特利尔议定书》,其中规定我国作为发展中国家在2040年之前可以生产和使用氟氯烃二氟一氯甲烷(R-22)作为制冷剂,发达国家早已经完成对R-22的淘汰,而我国R-22替代品的开发迫在眉睫.
HFOs由于其低的臭氧消耗潜能值(ODP)和全球变暖潜能值(GWP),在空气中较短的寿命,不会对大气中的臭氧层造成很大的破坏,成为R-22替代品的主要研究对象.迤今,生产HFOs的三大公司分别为霍尼韦尔公司、杜邦公司以及阿科玛公司.中国是世界上消耗臭氧层物质的生产和消费大国,但是生产的氟化工产品大都是廉价的初级产品.国内大多数企业生产的高附加值、高尖端产品均是与上述三大公司合作生产,目前只有浙江环氟新材料有限公司已经具备自主知识产权的百吨级中试生产含氟丙烯的能力,中化近代环保化工有限公司拥有自主研发的生产专利,但无规模化、商业化生产.其技术难点在于氟化催化剂的制备,以及目标产物的分离提纯,具有高选择性、高活性、高稳定性的催化剂是制备氢氟烯烃的关键.
合成HFOs的方法有很多种,根据其原料的不同,可以分为含氟烷烃的脱氟化氢、氢氟烯烃的加氢和脱氟化氢、氯氟烷烃的脱氯化氢、氯氟烯烃的脱氯化氢和氟化氢加成以及异构化反应等.气相合成HFOs所用的催化剂也有很多种,其中铬基催化剂是目前为止应用最广泛,催化性能较好的催化剂,也是本课题组目前首要的研究方向.本文简要介绍了合成HFOs的常见催化反应类型以及反应机制,介绍了各类合成氢氟烯烃催化剂的制备方法以及催化性能,并展望了该催化剂的未来发展方向.
1 合成氢氟烯烃的催化反应类型
在HFOs合成过程中,由于原料的不同会涉及到氟化反应、脱氟化氢反应、脱氯化氢反应等不同的反应类型.
1.1 氟氯交换反应
氟氯交换反应分为气相氟氯交换反应和液相氟氯交换反应.气相氟氯交换反应与液相反应相比更具优势,因为其产生的污染少、腐蚀少并且可以连续工作,是合成HFOs的主要途径.通常情况下是以HF为氟源在催化剂的表面进行亲核取代反应,催化剂主要为Cr基和Al基催化剂,主要是通过CCl2F2在催化剂表面的反应来研究氟氯交换反应的机理.Alonso等[9]制备了α-AlF3和γ-AlF32种高表面积的催化剂,通过CCl2F2和CHClF2在催化剂表面进行氟氯交换反应,探究氟氯交换反应机理.研究发现,α-AlF3具有更高的催化活性,归因于α-AlF3有更多的路易斯酸性位点,而结晶的α-AlF3对氟氯交换反应几乎没有催化活性.他们猜测催化剂表面具有氟原子和空位,CFC化合物可在一个电子空位(路易斯酸位)失去1个氯原子,并吸收1个氟原子.Bailey等[10]研究了CCl2F2在β-AlF3(100)上的吸附,认为氟氯交换反应是CCl2F2中的Cl原子吸附到Al的电子空位上,C—Cl键解离,与附近表面上的F离子形成新的C—F键,新形成的CClF3解吸后留下表面的Cl离子.下一个CCl2F2是通过C—F键吸附,进行C—F键解离,C和之前沉积的Cl离子之间形成一个键,继续进行新的CCl3F解离.
关于气相氟氯交换反应的机理尚不清楚,但是在有关机理研究中均表明了路易斯酸位点与催化剂表面不稳定的F离子是进行氟氯交换反应的关键.
1.2 脱卤化氢反应
卤代含氟烷烃气相脱卤化氢是目前合成HFOs的主要方法,脱掉的卤代烃主要为氯化氢或氟化氢.气相脱卤化氢所用的催化剂主要以活性炭、活性炭负载型催化剂、氟化物(Al,Cr和Mg)等为主.
吕剑等[11]认为催化剂的酸强度对稳定性和选择性均有巨大影响.活性组分的价态对脱卤化氢也有影响,三价Fe催化剂以及AlF3等金属氟化物具有更强的脱HF作用,而Li,Mg等一价、二价金属氟化物更适合脱HCl.Mao等[12]在1,1,1,2-四氟-2-氯丙烷气相选择性脱氯化氢制备2,3,3,3-四氟丙烯的反应中,研究了不同酸碱度催化剂表面的反应机制.在酸碱双功能催化剂上倾向于发生E2机理,而在含有不平衡强度的酸碱对的碱性催化剂上可能发生E1cB机理(Scheme 1).在E2机理中,碱性位点将质子从碳原子上拉开,同时氯原子从酸性位点与C—Cl键分离.在该反应中由于K和Cs改性MgO催化剂的表面化学硬度,导致高的脱氯化氢选择性.推测该反应可能以E1cB机理进行.这种机制通常发生在具有强相互作用的强碱性中心和弱酸性中心与卤化物相互作用的系统中.但是由于E2和E1cB机制的化学吸附在实验上难以区分,尽管进行了许多研究但脱卤化氢的真正机理仍不清楚.
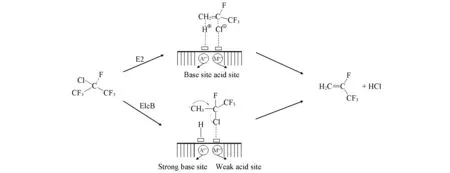
Scheme 1 Possible dehydrochlorination mechanisms on various Mg-based catalysts
1.3 催化加氢脱氯反应
氢氯氟烃化合物因为化合物中的氯会消耗大气层中的臭氧,而且随着氯含量的增加,臭氧消耗能力也随之增加.加氢脱氯反应可以对过剩的含氯化合物进行有效处理,使其转化为高附加值、无毒无害的产品.
加氢脱氯反应所用的催化剂主要是贵金属催化剂负载在活性炭上,贵金属可以吸附分子中的氯原子从而使C—Cl键解离,还可以有效地活化氢分子,受副产物HCl的影响较小.主要是通过CF2Cl2的加氢脱氯反应来研究反应机理.Morato等[13]最早系统地提出该反应的机理(Scheme 2),他们认为CCl2F2与催化剂表面的活性位吸附脱氯生成·CClF2自由基,加氢生成CHClF2或者继续脱氯生成·CF2自由基,·CF2自由基继续加氢生成CH2F2或者脱氟生成CH4.

Scheme 2 CF2Cl2hydrodechlorination reaction mechanism
Wiersma等[14]认为该反应机理是由2个平行的反应通道形成的(Scheme 3),CCl2F2与催化剂表面的活性位吸附脱氯生成·CClF2或者脱氟生成·CCl2F,自由基·CClF2加氢生成CHClF2或者继续脱氯生成·CF2自由基,·CF2自由基继续加氢生成CH2F2,自由基·CCl2F继续脱氟脱氯生成CH4.Zheng等[15]也证实了这些观点.从反应推测的各个机理中可以得出反应选择性关键在于自由基·CF2在催化剂表面的吸附速率,·CF2在催化剂表面脱附速率越高或者氢解速率越慢,CH4的选择性越低.

Scheme 3 CF2Cl2hydrodechlorination reaction channel
2 合成氢氟烯烃的催化剂
气相合成HFOs所用的催化剂有很多种类,按照主催化剂成分可以分为铬基催化剂、铝基催化剂、镁基催化剂和其它催化剂等,催化剂的制备方法也很多,以下介绍了各类催化剂以及催化剂的主要制备方法.
2.1 铬基催化剂
二十世纪九十年代,Tsuji等[16]就公布了铬基氟化催化剂,用于生产氢氟代烃(HFC)和氢氯氟代烃(HCFC),一般为三氧化二铬或者氟化铬为主催化剂,制备方法主要有共沉淀法、浸渍法、干混法,通过添加其它金属氧化物(Fe,Zn,Co等)进行改性,可以得到理想的转化率和收率.
2.1.1 沉淀法制备铬基催化剂 Brunte等[17]通过在硝酸铬溶液中加入氨水作沉淀剂,经过干燥焙烧后获得三氧化二铬催化剂.将获得的催化剂用于1,1,1-三氟-2-氯乙烷(HCFC-133a)的氟化反应中,并通过程序升温还原和氧化(TPR-TPO)对催化剂进行表征,探究了在氮气、氢气和空气气氛下预处理催化剂对催化活性的影响.研究发现,HCFC-133a氟化合成CF3CH2F的反应取决于催化剂中可逆的氧化铬物种,而在H2/O2摩尔比为2时,预处理的催化剂中可逆的氧化铬物种含量最多.
为了获得更高的活性和稳定性,Merkel等[18]在Cr2O3催化四氯丙烯和HF气相氟化合成2-氯-3,3,3-三氟丙烯中加入二异丙基胺等稳定剂,与没有稳定剂存在的反应相比,有效地提高了催化性能,延长了催化剂的寿命,并提高了反应物的转化率和目标产物收率.Tong等[19]基于氧化铬本身对气相氟化有很高的催化活性,直接用氟化的无定形氧化铬作催化剂,催化1,1,1,3,3-五氯丙烷合成1-氯-3,3,3-三氟丙烯的反应.通过将氮气、空气、氧气等与反应物一起放入反应器中,延长催化剂的寿命.研究发现,将氧气引入到氟化反应器中可以有效抑制催化剂表面的结碳行为,将催化剂的寿命延长两倍.
Mao等[20]通过在氟化氧化铬催化剂中添加Y或La等改性剂,将其用于1,1,2,3-四氯丙烯与氟化氢反应合成2-氯-3,3,3-三氟丙烯的反应中,获得较高的转化率和选择性.相比氟化氧化铬,经过La改性的氟化氧化铬催化剂能够在很长时间内保持高的转化率和选择性,并通过对反应机理的研究提出催化剂表面CrOxFy物种和BET比表面积与活性有关.La的加入可以促进CrOxFy活性物种的形成,从而提高催化活性.Wang等[21]制备了系列Y2O3-Cr2O3(F)催化剂用于1,1,2,3-四氯丙烯氟化合成2-氯-3,3,3-三氟丙烯,研究发现,在Cr2O3中掺杂钇可能会导致催化剂中高价铬物种的形成,在预氟化过程中可以转化为具有催化活性的CrOxFy物种,添加钇有助于保持对2-氯-3,3,3-三氟丙烯的高选择性.He等[22]通过沉积沉淀法制备系列的CrOx-Y2O3催化剂,用于2-氯-1,1,1-三氟乙烷氟化为1,1,1,2-四氯乙烷反应中.制备方法为:将Cr(NO3)3的水溶液与Y(OH)3粉末混合,然后在搅拌下将1 mol/L(NH4)2CO3的水溶液添加到混合物中直至出现沉淀获得浆液;将生成的浆液老化2 h,然后与母液分离,用去离子水洗涤,并在120℃下干燥过夜;最后,分别将其在400,500,600和800℃下煅烧4 h,探究煅烧温度对CrOx-Y2O3催化剂的影响,研究发现,在400℃下煅烧的预氟化催化剂具有最高活性,在320℃下CF3CH2Cl转化率为19%.随着煅烧温度升高,CrOx物种的结构从高度分散的单色铬酸酐(CrO3)转变为低氧化态的CrOx物种[Cr(Ⅴ)和Cr(Ⅲ)]和低聚铬酸盐.在活化过程中,部分分散良好的铬酸酐(CrO3)转化为催化活性物种CrFx,CrOxFy或Cr(OH)xFy.随着煅烧温度的升高,分散良好的铬酸酐含量降低,导致活性降低.Xie等[23]通过沉淀法制备Cr2O3催化剂,并用于2-氯-3,3,3三氟丙烯氟化合成2,3,3,3-四氟丙烯中,同样探究煅烧温度对催化活性的影响.结果表明,随着煅烧温度的升高,催化剂的微晶尺寸增加而表面酸性位点降低,在500℃下煅烧的催化剂能够表现出最高的活性,转化率达到63.3%.
Zhou等[24]在六氯丁二烯(HCBD)气相氟化合成1,2-二氯四氟环丁烯中,用沉淀法制备含有不同促进剂(Ni,Cu,In,Al)的CrOx/ZnO催化剂,研究发现,Cr-Ni-Zn催化剂使1,2-二氯四氟环丁烯(DTB)的收率达到90%.表征结果表明,Cr-Ni-Zn催化剂具有较大的比表面积、最大的孔径,更多的CrOxFy物种,最高的氧气浓度和广泛分布的酸强度.
Jia等[25]通过沉积沉淀法制备不同Cr2O3含量的Cr2O3-AlF3催化剂,用于氟化二氯二氟甲烷合成四氟甲烷反应.研究发现质量分数为61.2%Cr2O3的Cr2O3-AlF3催化剂具有最高的氟化活性.Zhou等[26]提出了一种含铟、钴、铬3种金属离子的三组分铬基催化剂,其中In与Cr的摩尔比在0.02~0.15的范围内,Co与Cr的摩尔比也在0.02~0.15的范围内.用于乙炔与无水氟化氢反应生成1,1-二氟乙烷,转化率达到75%以上,选择性达到90%以上.Lu等[27]提出了一种由1,1,2,3-四氯丙烯合成2-氯-3,3,3-三氟丙烯的催化剂及其制备方法,催化剂由Cr,Al和Zn,Co组成,在一定反应条件下,转化率为99.4%,收率为89.3%.Hu等[28]用沉淀法制备了Cr2O3催化剂,所不同的是在预氟化时,Cl2和O2作为氧化剂与无水氟化氢一起通入.研究发现,被氧化的催化剂具有更大的比表面积和更强的表面酸度,在HCFO-1233xf合成HFO-1234yf的反应中表现出优异的催化性能.
Luo等[29]通过向氧化铬里掺杂镍得到比Cr2O3更具活性的NiO/Cr2O3催化剂,研究发现,NiO/Cr2O3活性高的原因主要有2点:首先,NiO/Cr2O3的表面酸位密度较低,因此比Cr2O3更加稳定,可以缓解焦炭在催化剂上的沉积;其次,催化剂在氟化预处理过程中形成NiF2,导致反应新活性位点的产生;动力学研究还表明NiO/Cr2O3的活化能比Cr2O3的活化能低很多,意味着前者催化剂上的反应途径不同.因此,在NiO/Cr2O3催化剂上提出的反应机理涉及在NiF2表面进行额外的脱氟化氢反应.Ni2+阳离子充当酸性位点,而F-阴离子充当碱性位点,分别负责HFC-245fa分子中C—F和C—H键的活化和裂解.
本课题组[30]采用公开沉淀法制备Zn/Cr2O3三组分催化剂,主催化剂为三氧化二铬,助催化剂为氧化锌和活性炭,在460℃,H2气气氛下焙烧4 h后得到具有高活性、高选择性、高稳定性的氟化催化剂. 在1,1,1,3,3-五氯丙烷气相氟化合成1-氯-3,3,3-三氟丙烯的反应中,反应温度为200 ℃,氟化氢与1,1,1,3,3-五氯丙烷的摩尔比为10∶1时,转化率大于99%,选择性为98.2%,并且在100 h内没有失活.XRD结果[图1(A)]表明,在催化剂中大多数氧化铬以无定形的形式存在从而导致催化剂的高活性和稳定性.催化剂的比表面积越大催化剂的活性越高.XPS结果[图1(B)]表明,在预氟化过程中,催化剂表面的铬氧化物与氟原子发生强相互作用,形成活性中心.吡啶吸附红外光谱和氨气程序升温脱附结果证明,尚未失活的催化剂Lewis酸和Bronsted酸中心的数目和强度与新制备的催化剂相比明显提高.

Fig.1 XRD patterns(A)and XPS spectra(B)of the Cr2pfor the fresh catalyst Zn/Cr2O3(a)and catalyst Zn/Cr2O3after 118 h reaction(b)
2.1.2 浸渍法制备铬基催化剂 吕剑等[31]通过在Cr2O3上浸渍氯化铁,经过干燥、焙烧、活化后得到三价铁负载的Cr2O3催化剂,在1,1,2-三氯-3-氟丙烯氟化合成2-氯-3,3,3-三氟丙烯反应中,反应温度180 ℃,接触时间5 s,得到1,1,2-三氯-3-氟丙烯转化率为100%,2-氯-3,3,3-三氟丙烯选择性为99%.Mao等[32]通过在Cr2O3上浸渍Mg,Ca,La或Y来对催化剂进行改性,进行1,1,2,3-四氯丙烯氟化合成2-氯-3,3,3-三氟丙烯的反应.研究发现,通过在Cr2O3中添加Mg,Ca和La可以促进高度分散的CrOxFy物种的形成,而添加Y会抑制CrOxFy物种的形成.此外,向Cr2O3中添加La和Y可以增加氟化催化剂的比表面积.在Cr2O3催化剂中加入La可以使催化剂表现出最好的活性和寿命.
Wang等[33]通过浸渍法制备CrO3/Cr2O3催化剂,制备方法是用H2CrO4水溶液浸渍Cr2O3,然后在80℃下蒸发悬浮液,得到固体产物,在N2气气氛下于150℃干燥4 h,催化剂中的CrO3摩尔分数为10%.由于高价铬在预氟化过程中会形成活性物种CrOxFy,提高了催化剂的活性,但在反应过程中,CrOxFy活性物种转化成稳定但无活性的CrF3,从而导致催化剂的失活.
2.1.3 混合法制备铬基催化剂 张永明等[34]提出一种以乙炔为原料,经过氟化反应生成1,1,-二氟乙烷的反应,其中催化剂为活性炭与氯化铬、氯化钯的混合物,氯化铬为催化剂总量的10%~20%,氯化钯为催化剂总质量的3%~8%,反应温度300℃,乙炔与氟化氢体积比为1∶12,反应器压力为0.5 MPa时,乙炔的转化率高达98%,1,1,-二氟乙烷的选择性为99.5%以上.齐芳等[35]提出了一种高比表面积的氟化催化剂的制备方法,将铬盐和镧盐溶解到水中,加入乙二胺四乙酸(EDTA)和氨水,形成凝胶,将凝胶和少量氟化铝、石墨干混压片后于250℃焙烧,得到比表面积为450 m2/g的催化剂,用于HCC-240db氟化合成HCFC-1233xf的反应,得到高达98%的选择性.
2.1.4 其它方法制备铬基催化剂 由于铬基催化剂失活的主要原因是催化剂表面结碳,传统的制备方法不能够很好地解决这个问题,近年来通过制备方法创新对催化剂的结构进行调控从而提高了催化剂的活性和稳定性.Han等[36]通过CrCl3水溶液与NaBH4溶液反应,制备一种六方柱结构的Cr2O3,用于1,1-二氟乙烷生产氟乙烯.具体制备方法为:将CrCl3溶液缓慢滴加到NaBH4溶液中,并剧烈搅拌1 h,使CrCl3与NaBH4完全反应,获得蓝色溶液;通过离心分离蓝色溶液获得沉淀,将获得的沉淀用蒸馏水洗涤3次,并在60℃下干燥24 h;最后,将样品在N2气气氛中于500℃下煅烧2 h.表征结果表明,成功制备棱镜长为(285±43)nm,宽度为(233±33)nm的均匀Cr2O3六角棱镜结构,以直径小于3~5 nm的具有多晶结构的纳米Cr2O3的疏松状和网状聚集的形式存在,在催化剂表面具有相对丰富和强酸性位点.具体的反应过程如下:

与商用Cr2O3相比,Cr2O3晶体的尺寸小得多,具有较高的表面能,较低的强度和更丰富的路易斯酸度,有更高的活性和稳定性.由于这种六方柱结构的介孔结构使催化剂具有更高的比表面积,在反应70 h后没有发生明显的形态变化和催化剂烧结.
Han等[37]通过溶液燃烧法制备纳米氧化铬,用于1,1-二氟乙烷的脱氟化氢反应.以甘氨酸为燃料,Cr(NO3)3为Cr前驱体,通过溶液燃烧法合成相对均匀的薄片状催化剂.与商用Cr2O3催化剂相比,该方法制备催化剂表面上的CrO3含量高,预氟化后,催化活性和稳定性提高.Zhang等[38]采用硬模板法制备了有序的Cr2O3纳米棒,与沉淀法制备的Cr2O3颗粒相比,具有更高的面积比反应速率.研究发现,Cr2O3纳米棒具有更高的表面高价铬含量和表面酸性位点密度,是提高反应活性的2个关键因素.
Lim等[39]通过溶胶-凝胶法制备Cr2O3粉末,并在氟化氢水溶液中浸泡24 h,获得氟氧化铬.研究发现,由于表面氟氧化铬的形成,催化剂表现出良好的催化活性,氟氧化铬的活性可以通过氟化度来调节.Liu等[40]通过水热合成法制备MIL-101结构的,以Cr(NO3)3·9H2O为前体,以对苯二甲酸为有机配体的催化剂(Scheme 4),用于1,1,1,3,3-五氟丙烷脱氟化氢合成1,3,3,3-四氟丙烯的反应. 实验制备不同焙烧温度下准MIL-101结构的高浓度Cr2O3分子簇,XPS等表征结果证明温度越高其结构塌陷程度越高,研究了合适温度下催化剂的催化活性,筛选出煅烧温度为450℃时,Cr2O3分子簇的反应速率几乎是商用Cr2O3的4倍,并且没有明显失活.商用催化剂失活的主要原因是活性位点的积碳,但是由于Cr2O3分子簇为准MIL-101结构,活性位点分离,所以很大程度上避免了烯烃的聚合,提高了催化剂的活性和稳定性.

Scheme 4 Schematic illustration of the preparation of quasi MIL-101
Li等[41]通过金属有机框架(MOF)衍生的方法制备了系列基于氧化铬的催化剂,并应用于氯丁二烯的氟化反应中.研究发现,源自MOF的催化剂不仅具有高选择性,而且可以显著降低反应温度.由于MOF衍生的催化剂具有稳定的结构和抑制结碳的能力,与传统沉淀法制备的催化剂相比具有较长的使用寿命.此外,纳米MOF衍生的催化剂可以大大减少Cr的用量,这将有助于最大程度地减少Cr污染的风险.
2.2 铝基催化剂
铝基催化剂相对于铬基而言,具有对环境更加友好、价格便宜等优点,而且铝基催化剂有较强的酸性和大的比表面积,在催化领域中的应用十分广泛,所以近年来有关铝基催化剂的研究备受关注.大多数研究都是关于含氟烷烃的脱氟化氢反应,这是由于氧化铝独特的理化性质导致其成为HF的吸附剂,氟离子可以被氧化铝表面吸附,从而达到脱氟化氢的目的.铝基催化剂有以下几种制备方法.
2.2.1 沉淀法制备铝基催化剂 罗孟飞等[42]提出了一种用1,1,2,3-四氯丙烯和HF气相氟化反应合成2-氯-3,3,3-三氟丙烯和2,3-二氯-1,1-二氟丙烯的联产方法,Cr2O3-Al2O3催化剂中Al和Cr摩尔比为1∶1,在反应过程中,由于结碳的形成使催化剂的氟化性能下降,从而降低了2-氯-3,3,3-三氟丙烯的选择性,提高了2,3-二氯-1,1-二氟丙烯的选择性. 杨刚等[43]提出了一种由1,1,3-三氯丙烯或3,3,3-三氯丙烯制备2-氯-3,3,3-三氟丙烯的方法,催化剂为氟化铝类催化剂,将氢氧化铝粉末与氢氧化钠溶液混合后加入MgCl2和CuCl2,经过沉淀、洗涤、干燥、焙烧后得到氧氯化催化剂.反应温度350℃,原料CCl2CHCH2Cl/HCl/O2/HF摩尔比为1∶1.2∶2.4∶10,压力为0.3 MPa,空气流速为400 h−1时,转化率为99.9%,选择性73.54%.
Jia等[44]通过沉淀法制备具有不同酸度的多孔氧化铝催化剂,并进行1,1,1,2-四氟乙烷的脱氟化氢合成三氟乙烯反应.通过改变焙烧温度来有效地调节多孔氧化铝的路易斯酸度,多孔θ-Al2O3表现出优异的催化性能,转化率为35.1%,选择性为99.0%.研究发现,活性中心是适当数量的弱路易斯酸中心,而强路易斯酸中心可能导致催化剂快速失活.当路易斯酸位含量为0.76时,三氟乙烯的收率最高,而随着酸性位点的增加,三氟乙烯在催化剂表面聚合,催化剂快速失活.三氟乙烯是通过使用负载的Pd或Ru催化剂,对三氯三氟乙烷或三氟氯乙烯进行加氢脱氯而生成的,后期分离存在许多困难,且需要昂贵的贵金属.原始的CF3CFH2由于具有较高的全球变暖潜力,最终将被2,3,3,3-四氟丙烯替代.CF3CFH2向高附加值三氟乙烯的转化也解决了产能过剩的问题.
2.2.2 浸渍法制备铝基催化剂秦越等[45]通过浸渍法制备铝化合物负载型催化剂,将Al(NO3)3溶于水配成浸渍液浸渍AlF3-MgF2载体. 用于1,1,1,3,3-五氯丙烷氟化合成1-氯-3,3,3-三氟丙烯反应,在240℃,HF与1,1,1,3,3-五氯丙烷摩尔比为10∶1,接触时间5 s,转化率为100%,选择性为98.9%.Bonnet等[46]还提出了一种使用五氯丙烷气相氟化合成2-氯-3,3,3-三氟丙烯的方法,将浸渍镍和铬酐混合溶液的氟化氧化铝作为催化剂,向反应管中通入HF和N2气的混合气体,接触时间为7.4 s,HF与反应物摩尔比36,反应温度340℃,得到反应物的转化率为100%,选择性高达98.3%.此外,他们还提到可以延长催化剂寿命的阻聚剂,通过低含量(10-2~5×10-4)的阻聚剂(对甲氧基苯酚、叔戊基苯酚)共进料,可以有效地控制氯烯烃的聚合,延长催化剂的寿命.Swamidoss等[47]研究了分别在500和650℃煅烧的Al2O3和通过金属浸渍改性的催化剂Mg负载的Al2O3,在1,1,1,2-四氟乙烷脱氟化氢反应中的催化性能.研究发现催化剂强酸性位的数量随煅烧温度的升高而降低,而镁的加入增大了Al2O3催化剂的比表面积,并且使催化剂的弱酸性位增强,他们认为Al2O3的强酸位导致大量结碳,会缩短催化剂寿命,催化活性与弱酸性位点/强酸性位点的比率很好地相关.在650℃煅烧的催化剂上,弱碱性位点的增加和强碱性位点的减少也是提高Al2O3和Mg/Al2O3寿命的重要因素.
Jones等[48]研究由锌浸渍的γ-Al2O3组成的一系列催化剂,采用程序升温反应研究CCl4是通过顺序机制氟化的,其中CCl3F为主要产物,CCl2F2为次要产物.他们认为活性氟由不稳定的表面键合H—F组成,催化剂的表面进行氟化至多层深度,同时保留其缺陷尖晶石结构有利于提高其催化活性.
我们[49]以γ-Al2O3为载体,制备不同过渡金属离子溶液浸渍的负载型催化剂,研究发现,齿形γ-Al2O3载体浸渍氯化铬溶液后,反应温度为200℃,氟化氢与1,1,1,3,3-五氯丙烷的摩尔比为10∶1时,转化率达到100%,选择性为85.6%,并且在32 h内没有失活;而球形γ-Al2O3载体,在氯化铬浸渍液中加入适量的氯化锌,可以提高催化剂的选择性,延长催化剂的寿命.锌负载在含铬氧化铝上对提高HCFO-1233zd(E)的选择性有益.
2.2.3 其它方法制备铝基催化剂 Mao等[50]用不同铝源,乙二醇介导的溶胶-凝胶技术制备高比表面积的纳米级α-氟化铝,研究了它们在1,1,1,2-四氟乙烷的气相脱氟化氢中的性能.研究发现,所得氟化物的相组成、物质结构、酸性性质大不相同.廉价的无机铝通过溶胶-凝胶路线合成氟化铝前体,制得的α-AlF3具有高比表面积(高达170 m2/g)和强表面酸性,经过400℃的热处理也是如此.当使用Al2(SO4)3作为前体时,硫酸根离子抑制了氟化铝在煅烧过程中的结晶,导致AlF3的非晶相.分别使用异丙醇铝和氢氧化二乙酸铝制备具有β和烧绿石相的氟化铝.红外光谱(IR)、X射线衍射(XRD)和元素分析的表征结果显示,乙醇酸铝的形成可以有效地减少前体的水解并抑制颗粒团聚,形成纳米氟化铝.少量水解产生的残留氧以抵抗高温处理下的烧结是提高所制备氟化铝热稳定性的关键因素.由于路易斯酸含量高,衍生自AlOOH和Al2O3的α-AlF3表现出极高的催化活性,CF3CH2F转化率为24%~28%,CF2CHF选择性高于98%,远高于通过传统方法制备的氟化铝催化剂.Fang等[51]使用溶胶-凝胶法和不同的铝前体制备系列γ-AlF3复合催化剂,用于HFC-245fa脱氟化氢反应,研究发现,在制备过程中,由于有机配体的不完全热分解,在催化剂中形成预沉积的碳质物种,该物种阻塞强酸性位,使催化剂具有非常优异的催化剂稳定性.
Del等[52]提出了一种特定的卤代丙烷或卤代丙烯气相氟化合成2-氯-3,3,3-三氟丙烯的方法,其中氟化催化剂为AlF3或者氟化氧化铝,反应温度为350℃,HF与卤代丙烷或卤代丙烯的摩尔比为5∶1,接触时间为5 s时,2-氯-3,3,3-三氟丙烯的收率为86%. 此外,Jia等[53]在1,1,1,2-四氟乙烷的催化脱氟化氢合成三氟乙烯(CF2-CHF)的反应中使用三氧化二铝催化剂.通过掺杂系列碱、碱土、稀土和过渡金属氧化物对催化剂进行改性后发现,NiO/Al2O3和Ce2O3/Al2O3的活性优于母体γ-Al2O3,Cr2O3/Al2O3催化剂在400~450℃的低温范围内显示出高活性,但是Cr2O3和相关物质作为催化剂中的掺杂剂对环境和人体健康均不利.所以该研究内容主要放在了NiO/Al2O3上,通过对NiO/Al2O3的表征分析发现,路易斯酸位对脱氟化氢反应起着重要的作用,通过硝酸镍改性的γ-Al2O3增加了新的路易斯酸位点,使催化剂的催化活性大幅度提升.
Han等[54]以甘氨酸为燃料,通过溶液燃烧法制备了片状纳米级Cr/Al复合催化剂,经过氟化预处理后,Cr物种与Al物种紧密相互作用,形成非晶态结构.该相互作用提供了相对大量的缺陷,并且有利于活性物种的形成,提高路易斯酸度和催化活性,使得该复合催化剂在1,1-二氟乙烷脱氢反应中的反应速率比Cr和Al催化剂高30%~40%.
罗孟飞等[55]在1,1,2,3-四氯丙烯气相氟化合成2-氯-3,3,3-三氟丙烯的反应中,应用了氟化铝,并通过对反应产物和催化剂表征分析对反应机理做出推测,由于在反应初始催化剂活性高,反应产物以三氟丙烯为主,但随着催化剂活性降低,反应物中二氟丙烯、一氟丙烯的含量增多,三氟丙烯的含量减少.他们认为在反应初期,由于催化剂活性高,四氯丙烯可在催化剂表面进行多次的加成与消除反应生成三氟丙烯,但随着催化剂表面结碳程度增加,活性中心减少,四氯丙烯加成、消去反应减少,氟化程度低的副产物随之增多.
2.3 镁基催化剂
氟化镁具有耐腐蚀性,尤其是在含有氟化氢这种腐蚀性介质的反应中具有独特的优势,表现出优异的稳定性,可以作为氟氯交换反应、脱卤化氢反应、脱氢反应、异构化反应的催化剂.未经改性的氟化镁比表面积较小、路易斯酸位弱、作为催化剂催化活性弱、稳定性不高.
为了扩大其应用范围,有很多研究通过改变氟化镁的结构或者添加其它金属调变其性能.与氟化铝等铝基催化剂相比,氟化镁的路易斯酸较弱,很大程度上避免了活性位点结碳而导致失活的问题.氟化镁催化剂比表面积小,通过不同的制备方法可以增加比表面积,但是在高温下烧结的问题还是没有解决.显然,抑制MgF2的烧结并因此保持高的比表面积和酸度是关键的挑战.
Han等[56]通过静电纺丝法将纳米MgF2嵌入碳纳米纤维和电纺MgF2纳米纤维中,作为1,1,1-三氟乙烷脱氟化氢的高效催化剂.具体制备方法如Scheme 5所示,将Mg(CH3COO)2·4H2O溶解在15 mL N,N-二甲基酰胺(DMF)中.室温下剧烈搅拌,加入1.0 g聚偏氟乙烯(PVDF),1.25 g聚乙烯吡咯烷酮(PVP),加热至60℃,搅拌至PVDF和PVP完全溶解.在室温下,继续搅拌12 h,通过超声处理混合溶液,然后静置4~5 h,去除溶液中的气泡,获得棕黄色透明的静电纺丝溶胶.将溶胶置于15 mL注射器中进行静电纺丝,进料速度保持在1 mL/h,收集距离(注射器针头和收集器之间的距离)为18 cm,商业铝箔充当收集器,电纺丝的电压保持在18 kV,静电纺丝15 h后,得到了圆形纤维毡[Mg(CH3COO)2·4H2O/PVP/PVDF复合纤维].然后,分三步煅烧复合纤维:首先,将复合纤维在110℃下干燥以去除残留的DMF和吸附的水;其次,将复合纤维在空气气氛中于220℃预氧化5 h;最后,将样品置于管式炉中,在N2气气氛中以30 mL/min的流速将温度从室温升高至650℃,升温速率为1℃/min,在650°C下保持5 h后,将温度再次冷却至室温,并获得了嵌入碳纳米纤维中的纳米MgF2.由于极小的MgF2颗粒嵌入碳骨架中或被限制在纳米纤维中,因此,在450℃甚至500℃的脱氟化氢反应过程中未观察到明显烧结.

Scheme 5 Schematic illustration of the fabrication procedure for sub-nano MgF2embedded in carbon nanofibers and MgF2fibers by one-step electrospinning
Mao等[57]研究的Fe负载的中空纳米MgF2催化剂在苛刻的条件下可以表现出优异的催化活性,其和铬基催化剂相比有着相似的活性,但由于其中等强度的酸位和稳定的纳米结构,使其使用寿命延长.研究发现,空心纳米MgF2不仅显著提高了纳米材料的稳定性,还降低了其表面酸度,并且中空的纳米MgF2可以用作获得高分散Fe的有力载体.
秦越等[58]提出了一种Fe-Mg复合氧化物和碳酸镁的混合物作催化剂前驱体,经过焙烧预氟化后制得催化剂并用于含氯丙烯与HF气相反应,合成2-氯-3,3,3-三氟丙烯.在反应温度为260℃,HF与反应物摩尔比10∶1,接触时间为10 s时,1,1,2,3-四氯丙烯的转化率为100%,2-氯-3,3,3-三氟丙烯的选择性为99.5%,且催化剂的寿命大于100 h.
2.4 炭基催化剂等催化剂
除了铬基、铝基、镁基催化剂以外,炭基催化剂采用活性炭为载体,因多孔、大的比表面积、耐酸碱强度高和便宜易得等特性,在很多领域具有广阔的应用前景.作为催化剂的载体时,除了上述优点外,还可以对表面进行改性,如可以改变氧官能团的含量、引入其它元素(N)、嫁接官能团等.炭基催化剂的主要制备方法为浸渍法.毛伟等[59]提出一种用活性炭作载体,碱金属、稀土金属和高价金属氧化物等作活性组分的催化剂,制备方法为将活性组分对应的可溶性盐的水溶液浸渍活性炭载体6 h以上,再经过干燥、焙烧制得催化剂.研究发现当催化剂活性组成(质量分数)为3%Na-10%Ce-8%Fe/AC时,HCFC-243b的平均转化率为97%,HCFO-1233xf的选择性为96%.
Wang等[60]以尿素和蔗糖混合溶液为碳源和氮源,制备N掺杂有序介孔碳(NOMC).在1-氯-1,1-二氟乙烷(HCFC-142b,CH3CClF2)裂解为偏氟乙烯(VDF,CH2=CF2)中表现出稳定的活性.实验结果表明,NOMC的结构参数对VDF选择性有重要影响,碳表面的N含量对NOMC的催化性能有重要影响,NOMC催化剂的结构参数提高了催化剂的耐焦性.与工业生产的650~700℃的VDF相比,NOMC催化剂能在400 ℃下生产VDF.Yu等[61]以Ba(OH)2为前驱体、NH4F/NH4Cl作为F和Cl源进行固相反应制备了BaClxFy催化剂和BaF2催化剂并用于1-氯-1,1-二氟乙烷催化脱氯化氢为偏二氟乙烯.在350℃时促进HCFC-142b裂解为VDF.研究发现,与BaF2催化剂相比,BaClxFy催化剂具有更好的催化性能,这可能是因为在催化剂制备过程中少量Cl的存在会改变Ba的化学状态,进而改变HCFC-142b的C—Cl键的吸附和活化.此外,在制备BaClxFy时,由于NH4Cl的掺杂,使得BaClxFy的粒径变得更小更均匀,可能在催化性能中起主要作用.Wang等[62]通过固态研磨方法以不同的Ca/Ba摩尔比制备了CaBaFx复合材料,并将其用于1-氯-1,1-二氟乙烷热解为VDF的反应中,使得该反应在相对较低的温度(350℃)下进行.研究发现,Ca的掺杂使得催化剂形成更小的颗粒,而且根据结合能的变化,Ba在复合催化剂中将电子给予Ca,这种电子转移使催化剂表面酸度产生变化,表现出最低的表面弱酸性位点,有利于HCFC-142b的脱氯化氢作用.
吕剑等[63]在五氯丙烷和HF气相氟化合成2-氯-3,3,3-三氟丙烯中,使用的催化剂为由质量分数为60%的碳酸钙和40%的氢氧化铁混合均匀,压制成型,450℃焙烧之后,再于400℃氟化氢氟化之后制得.反应条件为氟化氢与有机物的摩尔比为15∶1,接触时间为10 s,反应温度为280℃,一步气相氟化制得2-氯-3,3,3-三氟丙烯,收率可达94%.此外,他们还以1,1,2-三氯-3-氟丙烯和HF为原料气相氟化合成2-氯-3,3,3-三氟丙烯[64],氟化催化剂由质量分数为40%的草酸钙和60%的氢氧化铁组成,再经过焙烧和预氟化,当反应温度240℃时,得到HCFC-1231的转化率为100%,HCFO-1233xf的选择性为98%,在反应温度为240℃,HF与反应物摩尔比为10∶1,接触时间为5 s时,得到HCFC-1231的转化率为100%,HCFO-1233xf的选择性为99.2%.
3 总结与展望
气相氟化反应在制冷剂的制备中占有非常重要的地位,氟化催化剂的研究大多集中在提高活性和延长寿命方面.目前工业应用以铬基催化剂为主,但是由于高价铬对环境和人体健康均有负面影响,探索经济、绿色、高效的氟化催化剂,避免铬及其化合物产生环境污染而开发非铬基的气相氟化催化剂意义重大.此外,气相氟化反应的基础研究对工业应用有着深远的指导意义.通过对气相氟化催化剂进行综述,对于未来氟化催化剂的开发有以下几点建议:通过对氟化机理的深入研究来指导催化剂的设计,延长非铬基催化剂的寿命,提高非铬基催化剂的活性;通过调控催化剂表面弱、中、强酸的酸性位点的分布,使反应可以持续高效进行,或通过对催化剂形貌结构的调控来抑制催化剂表面的结碳行为;开发安全环保且便宜易得的催化剂,提高氟化反应在工业应用中的经济实用竞争力.
猜你喜欢
化工管理(2022年13期)2022-12-02
浙江化工(2022年8期)2022-09-05
材料工程(2022年3期)2022-03-20
有机氟工业(2020年2期)2020-07-04
浙江化工(2019年9期)2019-01-21
中国特种设备安全(2018年12期)2018-03-15
中国卫生标准管理(2015年17期)2016-01-20
中国当代医药(2015年9期)2015-03-01
中国塑料(2014年12期)2014-10-17
有机氟工业(2014年3期)2014-06-05