环化及亚胺/酰胺部分氢化一锅法串联反应合成1,2,3,4-四氢喹喔啉
2020-10-16 05:42:16潘一骁李艳稳韩佳宏赵浩强丁相元徐立进范青华
高等学校化学学报 2020年10期
潘一骁,李艳稳,韩佳宏,赵浩强,冯 宇,丁相元,徐立进,范青华,时 茜
(1.中国人民大学化学系,北京 100872;2.中国科学院化学研究所,北京 100190;3.温州大学化学与材料工程学院,温州 325035)
1 Introduction
1,2,3,4-Tetrahydroquinoxaline moieties are widely found in numerous biologically active natural products and pharmaceuticals[1—10],and they are also known as promising dyes and cell adhesion agents[11—13].Consequently,various methods have been established to access these useful compounds[14—27].The selective reduction of the easily accessible quinoxalines represents one of the most convenient and straight forward routes to these heterocycles[28—32].Traditional reduction methods usually involve the use of stoichiometric metal hydrides or reactive metals as the reducing agents,and are often plagued with narrow substrate scope,low functional compatibility,poor chemoselectivity and the generation of copious amounts of toxic waste[33—37].To circumvent these problems,tremendous efforts have been devoted to the selective reduction of quinoxalines with cheap molecular hydrogen in the presence of homogeneous or heterogeneous catalysts,and significant advances have been made in the past several decades[38—66].Promising results have also been achieved in catalytic reduction of quinoxalines using formates[67—74],Hantzsch esters[75],hydrosilanes[76,77],water[78—80]and ammonia borane[81]as the hydrogen donor.However,most of these reports strongly rely on the priorly prepared and purified quinoxaline substrates,and the step-economical synthesis of tetrahydroquinoxalines via in situ generation of quinoxalines from readily available starting materials remains a challenge.In 2013,Beller et al.[59]disclosed the efficient preparation of chiral 2-phenyl tetrahydroquinoxaline directly from phenylglyoxal and 1,2-diaminobenzene following sequential cyclization/Fe-catalyzed catalytic asymmetric hydrogenation reaction,but only one example was given[Scheme 1(A),Path a].Later,Shi et al.[82]reported a similar tandem process involving cyclization and transfer hydrogenation for the step-economical preparation of chiral 2-aryl tetrahydroquinoxalines from aryl glyoxals and 1,2-diaminobenzenes[Scheme 1(A),Path b].Zhou et al.[83]demonstrated a B2(OH)4-mediated one-pot double reductive amination of 1,2-diaminobenzenes with 1,2-dicarbonyl compounds to give rise to 1,2,3,4-tetrahydroquinoxalines with water as both the solvent and proton donor[Scheme 1(A),Path c].It has long been known that 1,2,3,4-tetrahydroquinoxalines can be prepared via deoxygenative reduction of the amide moiety of 3,4-dihydroquinoxalin-2(1H)-ones[84—86],but the application of this transformation in organic synthesis is often hampered by the requirement of an overstoichiometric amount of expensive and sensitive reducing hydride reagent such as LiAlH4and borane.In this respect,our group[87]have recently reported the development of a one-pot tandem procedure involving cyclization and sequential hydrosilylation of imines and amides with polymethylhydrosiloxane(PMHS)under the catalysis of B(C6F5)3for the step-economical construction of 1,2,3,4-tetrahydroquinoxalines directly from readily available 1,2-diaminobenzenes and α-ketoesters[Scheme 1(B),Path a].However,the concomitant formation of large amounts of siloxane waste strongly reduces the atom efficiency of this process.Clearly,the use of inexpensive molecular hydrogen as the reducing agent would be the optimal choice in terms of atom economy and waste minimization.Although catalytic deoxygenative hydrogenation of amides into amines is rather challenging because of the low electrophilicity of the amide carbonyl carbon and the difficulty in the control of C—O bond cleavage selectivity,exciting achievements have been made in this field in the past several years[88—109].In particular,the groups of Cole-Hamilton[97,98],Klankermayer[99—101],Beller[102]and Zhou[103]reported the efficient Ru(Ⅲ)-catalyzed reduction of various amides into amines under relatively mild conditions.Given the general applications of these catalytic systems,we became attracted to exploring their performance in step-economical construc-
Scheme 1 Synthesis of tetrahydroquinoxalines via in situ generation of quinoxalines(A)and quinoxalinones(B)tion of 1,2,3,4-tetrahydroquinoxalines directly from 1,2-diaminobenzenes and α-ketoesters through a one-pot cascade cyclization/hydrogenation of imines and amides.
With our continuing interest in the transition-metal catalyzed reduction of N-heteroaryl compounds[110—117],we report herein that 2-substituted-1,2,3,4-tetrahydroquinoxalines can be prepared step-economically in good to excellent yields with high tolerance of functional groups via a tandem process combining cyclization of 1,2-diaminobenzenes and α-ketoesters and the following sequential hydrogenation of imines and deoxygenative hydrogenation of amides under Ru catalysis[Scheme 1(B),Path b].
2 Experimental
2.1 General Information
Unless otherwise noted,all commercially available chemicals including organic solvents were used as received from Aldrich,Acros or Strem without further purification.Nuclear magnetic resonance(1H NMR and13C NMR)spectra were recorded on a Bruker Model Avance DMX 400 Spectrometer(1H 400 MHz and13C 100.6 MHz,respectively,Bruker Daltonics.Inc).High resolution mass Spectra(HRMS)were collected on a APEX Ⅱ electrospray ionization-mass spectra(ESI-MS,Bruker Daltonics.Inc.),4,5-Dibromobenzene-1,2-diamine(1d)[118],4,5-dimethoxybenzene-1,2-diamine(1e)[119],and α-ketoesters[120,121]were prepared according to the previous reports.Ru(acac)3[Ruthenium(Ⅲ)acetylacetonate],TsOH·H2O(p-toluenesulfonic acid),Yb(OTf)3·H2O[Ytterbium(Ⅲ)trifluoromethanesulfonate hydrate],In(OTf)3[Indium(Ⅲ)trifluoromethanesulfonate],HBF4[50%(mass fraction)solution in water],methanesulfonic acid(MSA),bis(trifluoromethanesulfonyl)imide(HNTf2)and 1,2-dimethoxyethane(DME)were purchased from Acros(purity 99%).1,1,1-Tris(diphenylphosphinomethyl)ethane(Triphos)was purchased from Alfa(purity 97%).
2.2 General Procedure
2.2.1 Synthetic Route of the Target Compounds 33aa— 33na A 10.0 mL glass vial equipped with a magnetic stirrer bar was sequentially charged with 0.25 mmol of o-phenylenediamine(1a),0.3 mmol of 2-oxopropanoate(2a),3.0 mg(0.0075 mmol)of Ru(acac)3,9.0 mg(0.015 mmol)triphos,3.11 μL(0.025 mmol,50%,mass fraction)of HBF4,25.0 mg of 0.4 nm molecular sieve(MS)and 1.0 mL ofn-Bu2O.Then the reaction vial was placed into a 25 mL autoclave.The autoclave was closed,and the final pressure of the hydrogen gas was adjusted to 3 MPa after purging the autoclave with hydrogen gas several times.The reaction mixture was stirred at 130℃for 18 h.After cooling the autoclave to room temperature,the hydrogen gas was carefully released.Then 5.0 mL of saturated aqueous NaHCO3solution was added to the reaction mixture,which was extracted with EtOAc three times(5.0 mL each time).The combined organic phases were dried over anhydrous Na2SO4,then filtered and evaporated under reduced pressure.After the removal of volatile materials by rotary evaporation,the resultant mixture was purified by silica gel column chromatography using a mixture of EtOAc and hexane to give compound 3aa.Taking compound 3aa synthesis as an example,compounds 3ab—3na were synthesized according to this method.The physical and chemical properties and NMR data of compounds 3aa—3na are listed in Tables 1 and 2,respectively.The NMR spectrum is shown in Fig.S1—Fig.S78(see the Electronic Supplementary Material of this paper).The synthetic route is shown in Scheme 2.

Table 1 Appearance,melting points,yields and HRMS data for compounds 3aa—3na

Table 2 1H NMR and13C NMR data for compounds 3aa—3na

Continued

Scheme 2 Tandem synthesis of tetrahydroquinoxalines via catalytic hydrogenation
2.2.2 Synthetic Route of the Target Compounds 44ab— 44yaA 10.0 mL glass vial containing a magnetic stirring bar was sequentially charged with 0.25 mmol of compoudn 1a,0.30 mmol of compound 2a,3.0 mg(0.0075 mmol)of Ru(acac)3,9.0 mg(0.015 mmol)of triphos,25.0 mg of 0.4 nm MS and 1.0 mL ofn-Bu2O.Then the reaction vial was placed into a 25 mL autoclave.The autoclave was closed,and the final pressure of the hydrogen gas was adjusted to 2 MPa after purging the autoclave with hydrogen gas several times.The reaction mixture was stirred at 130℃ for 4 h.After cooling the autoclave to room temperature,the hydrogen gas was carefully released.Then the reaction mixture was concentrated by vacuum evaporation and the residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane to give the pure compound 4aa.Taking compound 4aa synthesis as an example,compounds 4ab—4ya were synthesized according to this method.The physical and chemical properties and NMR data of compounds 4aa—4ya are listed in Tables 3 and 4,respectively.The NMR spectrum is shown Fig.S79—Fig.S100(see the Electronic Supplementary Material of this paper).The synthetic route is shown in Scheme 3.

Table 3 Appearance,melting points,yields and HRMS data for compounds 4aa—4ya

Table 4 1H NMR and13C NMR data for compounds 4aa—4ya

Scheme 3 Catalytic synthesis of dihydroquinoxalin-2(1H)-ones
3 Results and Discussion
3.1 Optimization of Reaction Conditions
We commenced our investigations with the optimization conditions for the reaction of compound 1a with compound 2a under H2and Ru catalysis.We first examined the catalytic performance of the ruthenium catalyst generatedin situfrom triphos and Ru(acac)3in the absence of any co-catalyst under 4 MPa H2at 140 ℃ in tetrahydrofuran(THF).After 18 h,the reaction went to completion,and a mixture of the target product 2-methyl-1,2,3,4-tetrahydroquinoxaline(3aa,yield of 15%),3-methyl-3,4-dihydroquinoxalin-2(1H)-one(4aa,yield of 80%)and 3-methylquinoxalin-2(1H)-one(5aa,yield of 3%)was obtained(Table 5,Entry 1).This observation suggested that the current catalytic system worked well for the cyclization of compound 1a with compound 2a to give compound 5aa and for the following hydrogenation of the imine moiety of compound 5aa to give compound 4aa,but its capacity to catalyze the final deoxygenative hydrogenation of the amide moiety of compound 4aa into compound 3aa appeared to be problematic.Other ruthenium complexes,such as[Ru(2-methylallyl)2(COD)]and[Ru(triphos)(tmm)],were also evaluated in this reaction,but none of them performed as effectively as Ru(acac)3(Table 5,Entries 2 and 3).Considering the importance of co-catalyst in improving the catalytic efficiency of Ru/triphos-catalyzed hydrogenation of carboxylic acid derivatives[117—123]andN-alkylation of amines[124—131],we speculated that the introduction of catalytic amount of additive might improve the catalytic efficiency.

Table 5 Optimization of reaction conditionsa
Then the effect of some additives was examined(Table 5,Entries 4—12).Delightfully,a catalytic amount of HBF4(10%,molar fraction)was found to increase the yield of compound 3aa to 88%(Table 5,Entry 6).Further optimization revealed that replacing THF with other solvents such as 1,4-dioxane,toluene and 1,2-dimethoxyethane(DME)only resulted in lower yields of compound 3aa(Table 5,Entries 11—13),but a slightly better yield of 90%was obtained in n−Bu2O(Table 5,Entry 14).Moreover,when the H2pressure was decreased to 3 MPa,the yield of compound 3aa dropped slightly in THF(Table 5,Entry 15),but in n-Bu2O the catalyst was equally effective(Table 5,Entry 16).Obviously,n-Bu2O is the choice of solvent.Furthermore,when keeping the H2pressure at 3 MPa while adding MS to the reaction system,the yield of compound 3aa was improved to 92%(Table 5,Entry 17).Notably,decreasing the reaction temperature to 130 ℃led to an excellent yield of 96%(Table 5,Entry 18).Although Beller et al.have shown that Ru(acac)3/triphos/Yb(OTf)3·H2O could work well for hydrogenation of various amides into amines[102],it only resulted in the formation of product 3aa in 25%yield in our case(Table 5,Entry 19).The catalytic system consisting of[Ru(H)2(CO)(triphos)],TsOH·H2O and BF3·Et2O,reported to effectively catalyze deoxygenative hydrogenation of various amides by Zhou et al.[103],only gave the target product 3aa in 63%yield(Table 5,Entry 20).Finally,the conditions in Entry 18 were chosen as the optimal reaction conditions.
3.2 Substrate Scope
Having established the optimized reaction conditions,we then turned to evaluate the reactivity of different α-ketoesters with compound 1a.As shown in Table 6,α-alkyl substituted α-ketoesters(2b—2k)underwent smooth reaction with compound 1a to provide the expected 2-alkyl substituted tetrahydroquinoxalines(3ab—3ak)in good to excellent yields.No significant steric effect on the reaction outcome was observed,although the presence of sterically demanding substituents resulted in slightly decreased yields(3af,3ag,3ai).The presence of a CF3group did not hamper the reaction with the product 3ak obtained in 75%yield.Likewise,α-aryl substituted α-ketoesters(2l—2u)bearing either electron-donating or withdrawing groups on the aryl ring were all tolerated,delivering the corresponding 2-aryl substituted tetrahydroquinoxalines(3al—3au)in high yields.Notably,halide substituents,such as F(3ap),Cl(3aq)and Br(3ar),were compatible with the reaction conditions,thereby providing ample opportunities for further chemical diversification.Compound 2t with an ortho-substituted methyl group gave a slightly lower yield probably as a result of steric hindrance.Similarly,ethyl 2-(naphthalen-1-yl)-2-oxoacetate(2v)delivered the product 3av in 76%yield.It is noticed that ethyl(E)-2-oxo-4-phenylbut-3-enoate(2w)exhibited good reactivity to afford the product 3aw in 85%yield,and the sensitive C=C bond remained intact throughout the reaction.To demonstrate the practicability of this methodology,we tried a gram-scale reaction of compound 1a(10.0 mmol)with compound 2a(12.0 mmol),and the desired product 3aa was obtained in 89%yield.
In addition to α-ketoesters,we also investigated the reaction of compound 1a with diethyl oxalate(2x),dimethyl carbonate(2y)and 4-methylene-2-oxetanon(2z)under the optimized reaction conditions.Gratifyingly,the corresponding products 3ax—3az with different ring sizes were isolated in moderate to good yields.
We next investigated the substrate scope with respect to 1,2-diaminobenzenes under the optimized reaction conditions(Table 6).The symmetrical 1,2-arylenediamines(1b—1f)bearing electron-rich and electrondeficient substituents on the aryl ring also performed well,giving rise to the corresponding tetrahydroquinoxaline products(3ba—3fa)in high yields.The electronic nature of the substituents on the aryl ring seemed to affect their reactivity as substrates containing electron-deficient substituents were slightly less reactive(3ca,3da).The N1-substituted benzene-1,2-diamines(1g—1i)were also converted efficiently to give the products 3ga—3ia.Notably,for substrate 1i,the allyl moiety remained intact during the reaction.Delightfully,other diamine compounds,such as 2,3-diaminomaleonitrile(1j),trans-1,2-diphenylethane-1,2-diamine(1k),trans-cyclohexane-1,2-diamine(1l)andN,N′-dimethylethylenediamine(1m)also exhibited good reactivity,providing the target products(3ja—3ma)in 52%—72%yields.Interestingly,the sensitive C=C bond in product 3ja was well tolerated in the reaction.
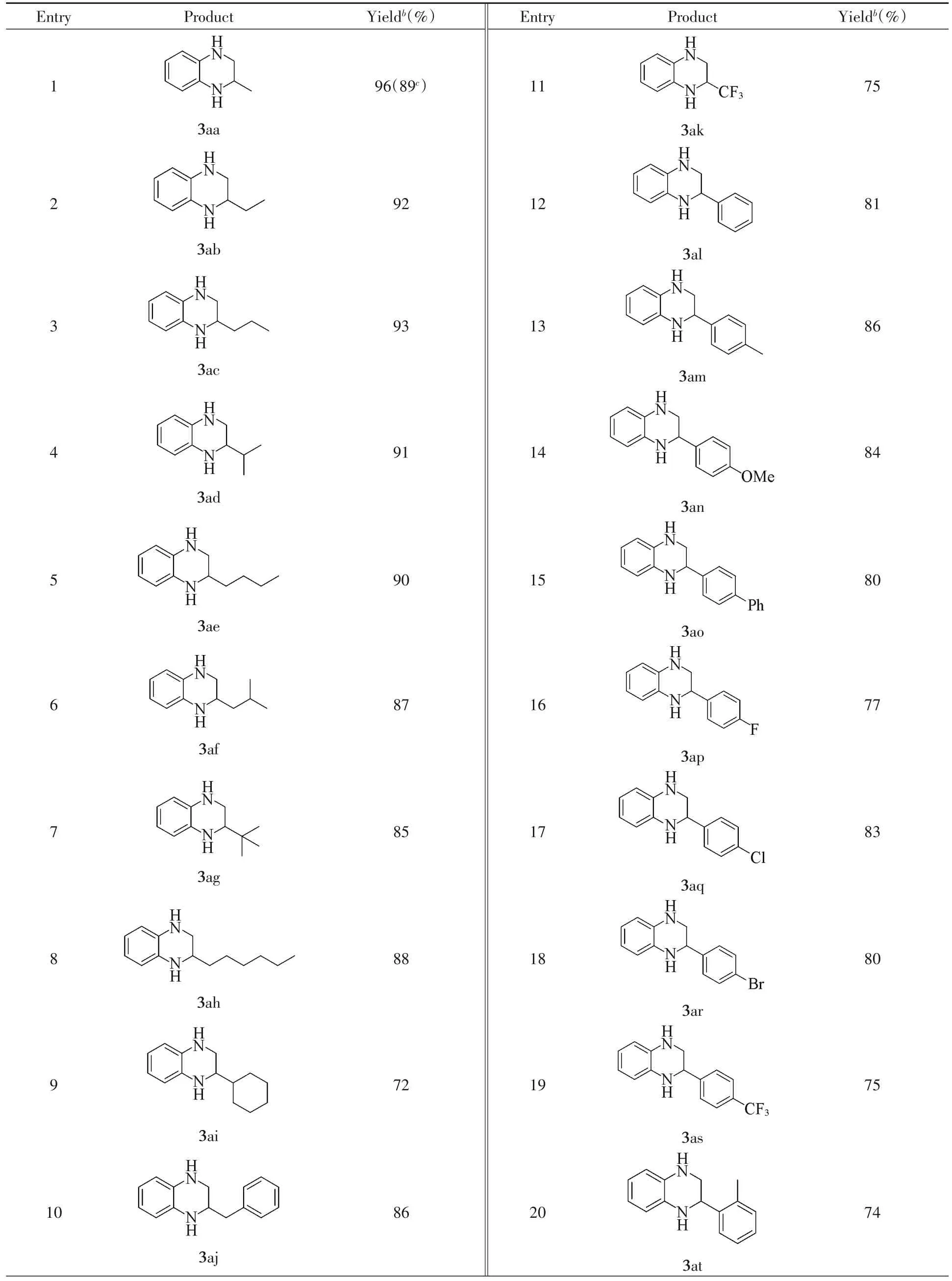
Table 6 Reaction scope with respect to α-ketoestersa

Continued
Notably,when using Ru(acac)3/triphos alone as the catalyst in the absence of any co-catalyst,the reaction of compound 1a and compound 2a provided 3-methyl-3,4-dihydroquinoxalin-2(1H)-one(4aa)predominantly in 83%isolated yield with a small amount of compound 3aa(10%)(Table 5,Entries 21).Delightfully,by lowering the hydrogen pressure to 2 MPa,compound 4aa could be exclusively obtained in 92%yield in 4 h,and the formation of compound 3aa was almost totally suppressed(Table 7).Similarly,the target products 4ab,4ad,4aj,4al,4aw and 4ba were isolated in excellent yields.The reaction of compound 1a with4-methylene-2-oxetanon(2y)led to the formation of product 4ay in 83%yield.2,3-Diaminomaleonitrile(1j)andtrans-cyclohexane-1,2-diamine(1l)gave the corresponding products 4ja and 4la in 85%and 69%yields,respectively.

Table 7 One-pot tandem synthesis of dihydroquinoxalin-2(1H)-onesa
To expand the scope of our catalytic systems further,the reaction of 2-aminophenol(1n)with compound 2a was also studied.As shown in Scheme 4,the synthetically useful compounds 3na and 4na could be selectively synthesized in good yields by tuning the reaction conditions.

Scheme 4 Ru-catalyzed tandem synthesis of oxazine and oxazin-2-one
Aiming to gain insight into the reaction mechanism,we first measured the kinetic profile of the reaction between compound 1a and 2a.As shown in Fig.1,almost all of compound 1a was converted into an intermediate identified as compound 5aa within 1 h by cyclization with compound 2a.Subsequent hydrogenation of compound 5aa led to the formation of compound 4aa,and this is followed by the deoxygenative hydrogenation of compound 4aa to give compound 3aa.Clearly,compounds 4aa and 5aa were intermediates during the reaction.HRMS analysis of the reaction system of compounds 1a and 2a under standard conditions revealed no formation of propane-1,2-diol.Thereby ruling out the possibility that the reaction proceeds via first reduction of the ketoester to the diol and subsequent alkylation.

Fig.1 Kinetic profile for the reaction of 1a and 2a under standard reaction conditions
To obtain more information about the reaction mechanism,the following several experiments were then carried out(Scheme 5).Full conversion of compound 1a into compound 5aa was observed in 1 h in the presence of molecular sieves without the use of Ru(acac)3/triphos catalyst,HBF4and H2,but a longer reaction time of 3 h was needed in the absence of molecular sieves[Scheme 5(A)].Clearly,the addition of molecular sieves was beneficial to the cyclization of compounds 1a and 2a,and the role of the in situ formed Ru(Ⅲ)catalyst was to catalyze the reduction of compounds 5aa and 4aa.Indeed,when subjecting compounds 4aa and 5aa to the standard reaction conditions respectively,the product 3aa was obtained in excellent yields in both cases[Scheme 5(B),5(C)].Further study showed that the reduction of 5aa in the absence of HBF4only resulted in the formation of compound 3aa in 15%yield,and compound 4aa was generated in 82%yield[Scheme 5(D)].These observations together with the results in Scheme 2 suggest that the Ru(acac)3/triphos catalyst alone could not work well for the reduction of compound 4aa into compound 3aa,and the presence of the HBF4cocatalyst is necessary for the smooth deoxygenative reduction of the amide carbonyl group.In order to understand the role of HBF4,a 4aa-HBF4adduct was isolated by heating a mixture of equivmolar compound 4aa and HBF4at reflux in ethanol,and its structure was determined by NMR spectra and HRMS analysis.This adduct could be hydrogenated under the catalysis of Ru(acac)3/triphos to deliver compound 3aa in 81%yield,with no need for additional HBF4[Scheme 5(E)].Notably,in the presence of catalytic amount of this salt,compound 4aa could be reduced with Ru(acac)3/triphos to afford compound 3aa in 89%yield,indicating the salt to be a catalytic active intermediate[Scheme 5(F)].This is in agreement with the previous studies about the application of Bronsted acids for amide activation in the transformations of amides in the literature[132].

Scheme 5 Experiments aimed to probe the reaction mechanism
Based on the aforementioned studies and previous reports[97—132],a reasonable reaction mechanism was suggested(Scheme 6).The starting materials compounds 1 and 2 initially undergoed cyclization to give the quinoxalinone intermediate 5.The following Ru-catalyzed hydrogenation of compound 5 led to the formation of dihydroquinoxalinone intermediate 4.Subsequent protonation of compound 4 with HBF4and enolization resulted in the formation of the zwitterionic adduct A,which was then hydrogenated into the intermediate B.The acid then promoted the dehydration of B to afford the iminium intermediate C.Finally the product 3 was produced upon hydrogenation of C under Ru(Ⅲ)catalysis.

Scheme 6 Proposed reaction mechanism
4 Conclusions
We have established an efficient,general and step-economical synthetic route to access a variety of 2-substituted 1,2,3,4-tetrahydroquinoxalinesviaa tandem one-pot process involving cyclization/hydrogenation/deoxygenative hydrogenation from readily available 1,2-diaminobenzenes andα-ketoesters under Ru(Ⅲ)catalysis.High yields,broad substrate scope and remarkable functional group compatibility were observed.The reaction was quite sensitive to the choice of co-catalyst,and HBF4was proved to be optimal.In addition,reducing the hydrogen pressure and omitting the co-catalyst led to the exclusive generation of 3,4-dihydroquinoxalin-2(1H)-ones in good yields.
Supporting Information:http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200333.
This paper is supported by the National Natural Science Foundation of China(No.21372258).
猜你喜欢
当代作家(2023年3期)2023-04-23 01:58:26
当代作家(2023年3期)2023-04-23 01:58:26
模具制造(2022年3期)2022-04-20 09:17:06
模具制造(2022年1期)2022-02-23 01:13:30
小读者(2021年4期)2021-11-24 10:49:03
考试与评价·高二版(2021年1期)2021-09-10 14:44:53
小天使·一年级语数英综合(2020年3期)2020-12-16 02:56:12
中国篆刻(2017年6期)2017-07-18 11:09:55
中国塑料(2015年6期)2015-11-13 03:02:55
中国当代医药(2015年7期)2015-03-01 02:01:21