
相互作用熵方法在计算生物分子体系结合自由能中的发展和应用
2020-10-16 10:27段莉莉黄开放钟素素
山东师范大学学报(自然科学版) 2020年3期
段莉莉 黄开放 钟素素
( 山东师范大学物理与电子科学学院,250358,济南 )
1 引 言
作为构成生物的基础物质,生物分子之间的相互作用在许多生命过程中起着核心作用.其中,蛋白质-蛋白质和蛋白质-小分子等分子之间的相互作用在控制基因表达,免疫反应,信号转导和酶抑制等许多生物过程都起着重要作用.生命大分子一般由简单的生物单分子重复聚合而成,如蛋白质由大量氨基酸脱水缩合而成,核酸由核苷酸聚合而成.而对生物大分子中相互作用的研究对于人们理解生命机理提供了可靠和直接的途径.研究它们之间的相互作用机制是疾病治疗和药物研制的基础.正常的生理活动都是一个动态过程,这意味着其中的生物大分子不仅整体都在运动,其内部也存在着相对运动,这给人们的研究带来了极大的挑战.尽管对生物大分子的相互作用研究面临着重重困难,但是这更激起了研究者的兴趣,人们在这一领域的研究也已取得了瞩目的进展.
通过X-ray表征生物大分子的三维结构为人们的研究奠定了基础,这使得研究者能够直观的感受生物大分子的空间结构[1, 2].后来居上的核磁共振(Nuclear Magnetic Resonance, NMR)技术使得人们能更准确细致的获取生物大分子的原子空间排布信息[3, 4].这些技术能将生物大分子表征出高分辨的三维结构,为研究者在原子层面上理解生物大分子之间的相互作用提供了化学基础.但是这些实验技术仅能提供生物大分子的静态空间结构信息,很难对其中的相互作用进行进一步的研究;同时,高昂的实验成本和漫长的实验过程也增加了这些实验方法的局限性.因此,采用理论方法来模拟生物大分子中的动态性质,对于理解其中的相互作用机理至关重要.
衡量生物大分子中的相互作用的强弱一般用结合自由能来描述,研究者根据不同的需要提出不同的理论方法用于结合自由能的计算.目前理论最严格和计算最精确的方法应属自由能微扰(Free Energy Perturbation,FEP)[5, 6]和热力学积分(Thermodynamic Integration,TI)[7, 8],其计算准确性已经在一些应用中得到了验证[9-27].这两种方法存在的主要的缺陷是结合自由能在计算时收敛得非常慢,这就导致其需要巨大的计算资源[28].尽管近些年已经开发出使用GPU进行FEP和TI计算结合自由能的算法[29, 30],但是这两种方法依旧不适用在大规模的生物大分子体系中.在计算速度上占据着绝对优势的打分函数在药物设计中虚拟筛选阶段被广泛使用,但是由于其较低的准确性一直被研究者谨慎对待[31-35].
1977年提出的分子动力学(Molecular Dynamics,MD)模拟运用势能函数描述生物大分子中原子之间的相互作用,然后由牛顿运动方程求解原子的运动状态,得到体系中每个原子的速度与位置随时间演化的信息,成功重现了分子实际运动的动态微观过程,为研究生物大分子构象变化的详细信息以及系统的热动力学性质提供了基础[36].MD模拟不仅有助于人们在原子水平上理解生物大分子体系的动态物理过程,而且还可以揭示在实验中难以检测甚至不能检测的构象.Kollman等人[37-39]借助MD能模拟出大量生物大分子构象的优势基础上发展了分子力学结合连续性溶剂模型(Molecular Mechanics/Poisson-Boltzmann Surface Area,MM/PBSA; Molecular Mechanics/Generalized Born Surface Area,MM/GBSA)结合自由能计算方法,该方法在计算精度和计算成本上做到了很好的平衡,因此被广泛地用于蛋白-蛋白、蛋白-配体、DNA-蛋白、RNA-蛋白和凝集素-糖之间的结合自由能的计算中.
MM/PB(GB)SA计算结合自由能的思想如图1.路径1描述的是实际中受体和配体中结合的方式:两者处于液态环境中并结合成复合物,其中ΔGsolv,bind为我们所求的结合自由能.考虑到在液相中两者结合的过程异常复杂,MM/PB(GB)SA模拟出路径2用以简化结合过程:配体与受体先在气相中结合,然后将结合后的复合物置于液体中.从图中可以看出.两者的路径始末状态相同,则图中的能量关系如下
(ΔGSolv,Rec+ΔGSolv,Lig)+ΔGsolv,bind=ΔGGas,bind+ΔGSolv,Comp
(1)
式中ΔGsolv,Rec、ΔGsolv,Lig和ΔGsolv,comp分别为气象中的受体、配体和复合物进入液相中时溶剂化能的变化;ΔGsolv,bind和ΔGGas,bind分别为液相中气相中的结合自由能.上式变形即可得结合自由能的公式
ΔGsolv,bind=ΔGGas,bind+ΔGSolv,Comp-(ΔGSolv,Rec+ΔGSolv,Lig)
(2)
通过该热力学循环可以巧妙地避开液态环境中复杂的能量变化,使得生物大分子体系中的结合自由能的计算变得可行且简便,这也正是MM/PB(GB)SA的优势所在.
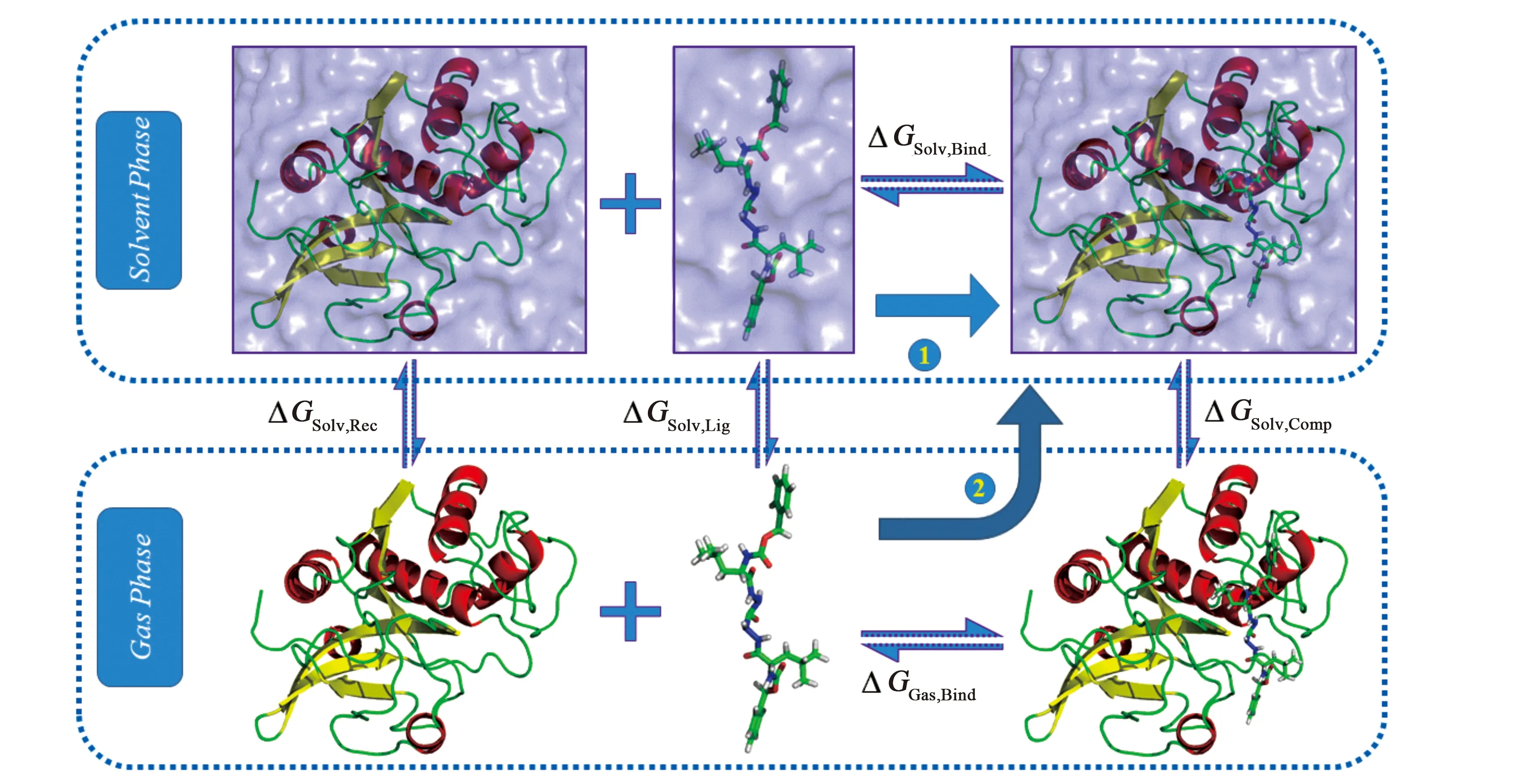
图1 MM/PB(GB)SA热力学循环图
在使用MM/PB(GB)SA计算结合自由能时,MD模拟过程中所使用的力场起着关键性的作用[40].张增辉等人[41]在2008年提出的蛋白质专一性极化电荷(Polarized Protein-specific Charge, PPC)力场,考虑了在传统力场中忽略的重要的静电极化效应,在计算蛋白质-蛋白质及蛋白质-配体小分子的相互作用中取得了突破性的进展,同时该方法也成功的折叠了一系列的蛋白,提高了传统力场在研究蛋白质折叠的效率[42-47].Weis等人[48]比较了AMBER ff94/99/03力场对于MM/PBSA影响,研究表明三个力场对计算准确性的影响相似.侯廷军等人[49]对AMBER ff99/99SB/99SB-ILDN/03/12SB力场研究后建议在MD模拟时和MM/PB(GB)SA计算时应使用相同的力场,而不应出现混搭情况.
通过MD模拟获得了系统大量的构象采样,然后进行结合自由能计算.目前MM/PB(GB)SA方法主要面临两个问题.其中一方面由于其本身参数化问题会导致计算结果与实验结果有一定的偏差.侯廷军等人[50]在2011年对MM/PB(GB)SA方法的性能进行了系统的评估,他们在研究中发现分子动力学模拟的时长、不同的内部介电常数以及不同的GB模型等,都对计算的结果有着敏感的影响.此后,他们通过对1 864个体系的测试发现MM/PB(GB)SA预测得到的结合亲和力往往比实验值更大,在某些体系中,采用较高的介电常数(如2或4)可以得到更好的结果[51].张增辉等人[52-54]通过测试大量实际体系中的不同介电常数对结合自由能的影响,发现对不同类型的氨基酸使用不同介电常数可以显著提高计算结果的准确性.
另一方面,在MM/PB(GB)SA的计算中,熵贡献通常采用传统的正则模(Normal Mode, Nmode)分析方法来计算[55-58].但是此方法存在的一个弊端是采用谐振子近似来估算复合物结合过程中熵的变化.由于该理论将熵的贡献分为平动熵、转动熵以及振动熵,因此忽略了非谐贡献,并且该方法计算量比较大[59-62].所以也无法适用特大分子体系中,因此许多研究者选择忽略了熵的贡献,这也导致计算结果更加不准确.为了解决目前熵变计算的困难,我们和张增辉教授课题组合作在2016年提出了相互作用熵方法(Interaction Entropy, IE)[63].该方法从MD模拟的轨迹中直接计算熵变,通过相互作用能的波动来衡量熵对结合自由能的贡献.与Nmode相比,该理论有着如下的优势:IE方法是通过严格的理论推导而得到,并且IE的计算能遍历MD中所有的构型,而Nmode一般只会抽样整个相空间的几十帧用于熵的计算,其次,尽管IE计算的构象要比Nmode多上数百倍,但是计算成本却远远低于Nmode,这是由于其直接对每一帧构象的“气相”中的相互作用进行计算,从而没有多余附加的而计算.最后,该方法还为研究者理解熵对生物大分子中的相互作用的贡献提供新的概念.鉴于该方法有诸多优势,IE的提出到现在也不过四年,但是研究者采用该方法去研究生物大分子的相互作用却如雨后春笋.
2 相互作用熵在具体体系中的应用
2.1周期蛋白依赖性激酶2(Cyclin-dependentKinases2;CDK2)与配体相互作用机制的研究CDK2属于CDK丝氨酸/苏氨酸激酶家族,其主要生理功能是调控细胞周期中G1期至S期的转换过程.CDK2结合Cyclin E或A后会磷酸化其Thr160残基,从而通过磷酸化其底物促进上述周期转换进程.CDK2的调控失衡会导致一系列后果,比如使细胞直接进入S或M期,从而导致细胞不受控分裂,即癌变.最近,一些研究发现CDK2也会参与细胞的分化当中[64].CDK2是肿瘤药物设计的前景靶标,因而一直是人们研究的热点.近些年,研究者发现水分子在蛋白质-配体之间的相互作用扮演者重要的角色,它能分别与蛋白质和配体形成氢键网络从而加强蛋白质与配体结合的稳定性[65].对于CDK2蛋白而言,其中的桥梁水分子对于蛋白质和抑制剂的调控作用往往被忽略.但是通过对众多的CDK2复合物的晶体结构观察发现,如图2所示,该复合物中往往存在一个水分子(WAT145),它既可以与CDK2的Asp145等残基形成氢键,同时又能与抑制剂形成氢键,起到“桥梁”作用.现有的一些对于CDK2的分子动力学的研究都是基于非极化力场,这也许会导致因忽略CDK2、配体、水分子的极化效应而产生误差.因此,极化作用和桥梁水的考虑将会对理解CDK2与抑制剂之间的相互作用提供一个新的视野.我们分别采用了蛋白质专一性极化电荷PPC和非极化力场AMBER研究了五个CDK2-配体的体系,同时针对桥梁水WAT145的影响进行了定量的分析[66].结果发现使用PPC力场进行动力学模拟,然后采用MM/PBSA结合IE方法计算它们之间的结合自由能能够极大提高计算结果的精度,尤其是在考虑桥接水分子情况下,计算结果与实验值的相关性达到0.98.而采用传统的MM/PBSA结合Nmode方法计算结果与实验值的相关性显著降低.该项工作指出,CDK2中的Ile10、Val18、Lys33、Glu81、Phe82、Leu83、Leu134及Asp145残基对桥接水分子、配体及CDK2激酶之间的结合起了非常重要的作用,桥接水分子WAT145与蛋白之间形成的氢键要比与配体之间形成的氢键更为稳定.这些关键残基与WAT145以及配体的相对位置如图2所示.WAT145桥水的存在会极大的增强CDK2-配体之间的结合亲和力,因此,该工作建议在药物设计当中可以设计出类似与WAT145的官能团以增强蛋白-配体的结合[66].

图2 CDK2体系晶体结构图、桥水和关键残基结构图
2.2蛋白内部介电常数对HIV-1蛋白酶与配体相互作用的影响获得性人类免疫缺陷综合症(Acquired Immune Deficiency Syndrome,AIDS)作为一种严重危害生命的传染病,在近些年来呈逐年增多的趋势.该疾病是由于感染人免疫缺陷病毒(Human Immunodeficiency Virus,HIV)引起的.HIV蛋白酶是一种逆转录酶,其包含HIV-1和HIV-2两种亚型.相比较而言,HIV-1因其传播广范而被人们广泛研究.HIV-1一经进入人体后就能快速攻击淋巴组织,感染其中的CD4+T淋巴细胞和巨噬细胞,从而使得人体丧失免疫功能.在计算HIV-1与抑制剂结合的复合物中,由于对溶质的介电常数无法进行精确的描述,所以导致MM/PB(GB)SA计算的结合自由能往往被高估.有研究表明,HIV-1的复合物中结合自由能的预测准确性要比其它复合物的要低,其主要原因是这些复合物与蛋白和抑制剂各形成两根氢键的桥梁水WAT301和未计算熵的贡献[67, 68].另外,其他的一些研究者在对HIV-1的研究中也遇到了计算结果不理想的状况[69, 70].
为了更可靠的探究HIV-1蛋白酶与配体相互作用,我们分别在PPC力场和AMBER力场下研究了蛋白质内部介电常数对HIV-1与配体的结合自由能预测的影响[71].同时,由于桥梁水WAT301存在于大多数的HIV-1体系中,因此该桥梁水对于HIV-1结合配体的影响也在本文中被研究.该项工作表明,在计算结合自由能只考虑焓变的贡献时,无论对于非极化的AMBER力场还是极化的PPC力场而言,改变蛋白质内部不同的介电常数不会对HIV-1与配体之间的结合自由能有太大的突破.尽管使用IE方法较Nmode能对预测结合自由能产生积极的影响,但当采用默认的介电常数1的时候所计算的结合自由能与实验值的相关性仍低于0.6.研究发现采用较大的介电系数能提高计算的精度.当内部介电常数设置为1.4~1.6、使用MM/PBSA计算焓变以及IE方法计算熵时,所预测的结合自由能与实验值的相关性达到0.84左右,且此时与实验值的MAE也是处于最小.当介电常数设置为1.8~2.0,MM/GBSA预测能得到较为理想的结果.该项工作还表明,当考虑WAT301水分子贡献时,能明显提高预测HIV-1与配体的结合自由能的准确性[71].
2.3利用IE方法和MM/PBSA方法研究间变性淋巴瘤激酶突变后的抗药性间变性淋巴瘤激酶(Anaplastic Lymphoma Kinase,ALK)隶属于胰岛素受体超家族,是一个受体型蛋白质络氨酸磷酸激酶.它调控着细胞的生长、分化、存活以及转化[72].由于其在众多的癌变中表现出异常的活性,因此ALK已成为一个治疗癌症的药物靶点.ALK由N末端、C末端和铰链环组成.在N末端中,存在CYS1156残基连接着β-折叠和α-螺旋;而在包裹抑制剂的铰链环中存在一个LEU残基.一些研究表明C1156Y与L1198F的突变极大影响着ALK与配体的相互作用,从而导致不同程度的耐药性[73-76].在过去的数十年中,由于ALK的变异已影响了三代抑制剂对肺癌的治疗,研究发现对于5P8抑制剂,L1198F单变异以及C1156Y/L1198F双变异已经产生了明显的耐药性;尽管C1156Y单变异能很好的被5P8治疗,但是却对VGH抑制剂有耐药性;VGH对L1198F单变异有甚于野生态的治疗效果.在与C1156Y有耐药性的相互抵消下,VGH抑制剂对于C1156Y/L1198F双变异体系有着和野生态几乎相同的抑制效果[73-77].
基于这些复杂的耐药机制,我们利用PPC力场和相互作用熵方法来研究ALK突变产生抗药机制的原因[78].研究表明,在PPC力场下,ALK与配体之间形成氢键的稳定性都高于非极化的AMBER力场.当发生突变时,ALK与配体的氢键稳定性会减弱.C1156Y变异造成结合自由能增强的主要原因是其增强了ALK与5P8之间的vdW贡献.尽管L1198F单变异也会导致复合物的vdW贡献增强,但是其总的极性贡献却减弱的非常明显.这导致当同时发生C1156Y和L1198F变异时,总的结合自由能减小.当然双变异后复合物的熵也有所增加是使耐药性增强的另外一个原因.对于另一种抑制剂VGH,C1156Y产生耐药性是由于vdW项、静电项的贡献减弱且熵变大引起的.但是VGH却能较理想的解决由L1198F单变异产生的抗药性,这是由于L1198F变异加强了PHE1198和GLU197与VGH之间的vdW相互作用.当产生双变异时,C1156Y变异产生的不利影响会与L1198F产生的有利影响相互抵消,这也解释了实际中为何VGH治疗C1156Y/L1198F双变效果与野生态类似.该工作从最基本的相互作用力出发,诠释C1156Y、L1198F和C1156Y/L1198F产生耐药机制的原理,不仅给研究者治疗ALK的耐药机制提供了理论基础,而且也对其它类似由变异产生的耐药机制提供了一种可行准确的研究方法[78].
2.4相互作用熵在凝血酶蛋白-配体相互作用中的研究凝血酶是一种在凝血级联反应结束时起着重要作用的丝氨酸蛋白酶.它主要负责在机体受到损伤时,将血浆中的可溶性纤维蛋白原转变成不溶的纤维蛋白,然后通过纤维蛋白聚合形成的纤维网络将血小板包裹起来,导致凝块的形成和血流的停止[79-83].然而,一些重要的医学疾病,如心肌缺血、脑血栓形成和心肌梗死等都与凝血酶有关.血栓栓塞性疾病是发达国家发病率和死亡率的主要来源,并且近年来发病率有上升的趋势.有限的疗效和出血性副作用减少了治疗这些疾病的可用药物的范围.因此,寻找有效的凝血酶抑制剂是科研人员和医务工作者面临的一项艰巨而有意义的任务.
考虑到在以往对凝血酶体系的研究中,极化效应一直被忽略,熵变也因其计算的困难往往被忽略.我们对10个分别在凝血酶抑制剂的P2和P3侧链进行修饰的凝血酶-小分子体系进行了结合机制的研究[84].研究结果表明,PPC力场结合新型的IE方法计算的结果与实验值更加一致,相关性达到了0.91.相反地,无论在传统的Amber力场下还是在极化的PPC力场下,通过Nmode计算熵变得到的相关性普遍较低.进一步对结合自由能的计算发现,凝血酶与小分子的结合主要来自于静电相互作用和范德华相互作用,其中Leu 96与小分子之间的CH-π和CH-CH相互作用与侧链的修饰引起的能量变化密切相关,Asp 199与配体之间的形成的两条稳定的氢键也提供了很强的静电相互作用.在结构方面,通过小分子的B-factor,蛋白质的主成分分析以及结合腔体积大小的分析来看,更加复杂的P2或P3侧链都增强了小分子与蛋白之间的相互作用,并且侧链中形成的环状结构与蛋白质结合的更加稳定.这项研究表明,IE方法在预测结合自由能方面优于传统的Nmode方法,强调了静电极化效应在分子动力学模拟中的重要性,同时揭示了不同结合位点的修饰配体结合自由能变化的原因,为更加有效的凝血酶抑制剂的设计提供了指导[84].
2.5MDMX/MDM2-p53/pDIQ相互作用的研究MDMX(Murine Double Minute X,Hdm4)与MDM2(Murine Double Minute 2,Hdm2)的过度异常表达会导致癌变.p53作为重要的肿瘤抑制因子和转录因子,直接参与了细胞的周期调控、代谢、生长、凋亡以及应激过程.MDMX/MDM2的过度表达会使p53的功能丧失,因此研究MDMX/MDM2与p53的相互作用对于治疗癌症是一个非常重要的课题.p53-MDMX/MDM2 相互作用是肿瘤药物设计的重要靶标,为了平衡MDMX/MDM2的过度表达,Phan等人设计出了与之结合后具有纳摩尔级的高结合亲和力的肽抑制剂pDIQ[85].pDIQ能抑制和破坏MDMX/MDM2与p53结合,从而维持p53的活性.实验表明,pDIQ-MDMX/MDM2之间能形成比p53-MDMX/MDM2更强的相互作用,同时MDM2与p53/pDIQ形成的相互作用也比MDMX与p53/pDIQ的强[85-87].
为研究MDMX/MDM2-p53/pDIQ相互作用的机制,采用IE方法对这四个体系进行了系统的研究[88].研究表明:相较于MDMX,p53/pDIQ与MDM2结合能产生更强的结合效果;相较于p53,pDIQ能与MDMX/MDM2结合的更为紧密.对于p53与MDM2结合比MDMX更强的原因在于MD模拟中前者体系比后者有更多的氢键形成.另一个原因是p53与MDM2之间的vdW相互作用要强于与MDMX之间.而pDIQ-MDM2体系的相互作用能强于pDIQ-MDMX的主要原因是vdW项的贡献更强.当与MDMX结合时,p53与pDIQ展现相同的热点残基,但是pDIQ的热点残基所贡献的能量总和要高于p53,这是pDIQ比p53能与MDMX结合更强的主要原因.对于MDM2,pDIQ较p53的结合更紧密的原因是pDIQ-MDM2体系中存在着不同于p53-MDM2体系的热点残基PHE55、VAL93、ARG65和LYS70,这些可能是导致p53-MDM2和pDIQ-MDM2之间的结合亲和力差异的主要因素.p53-MDM2 与pDIQ-MDM2体系的差异残基相对位置见图3.这项研究不仅阐明了MDMX/MDM2-p53/pDIQ相互作用的机制,还解释在实验中观察到pDIQ-MDM2结合亲和力最强、p53-MDMX最弱,p53-MDM2/pDIQ-MDM2处于中间相互作用机理.它不仅使得研究者能在原子层面上获知p53与MDMX/MDM2作用机理,还为针对MDMX/MDM2瘤药物设计的靶标提供了理论支撑.
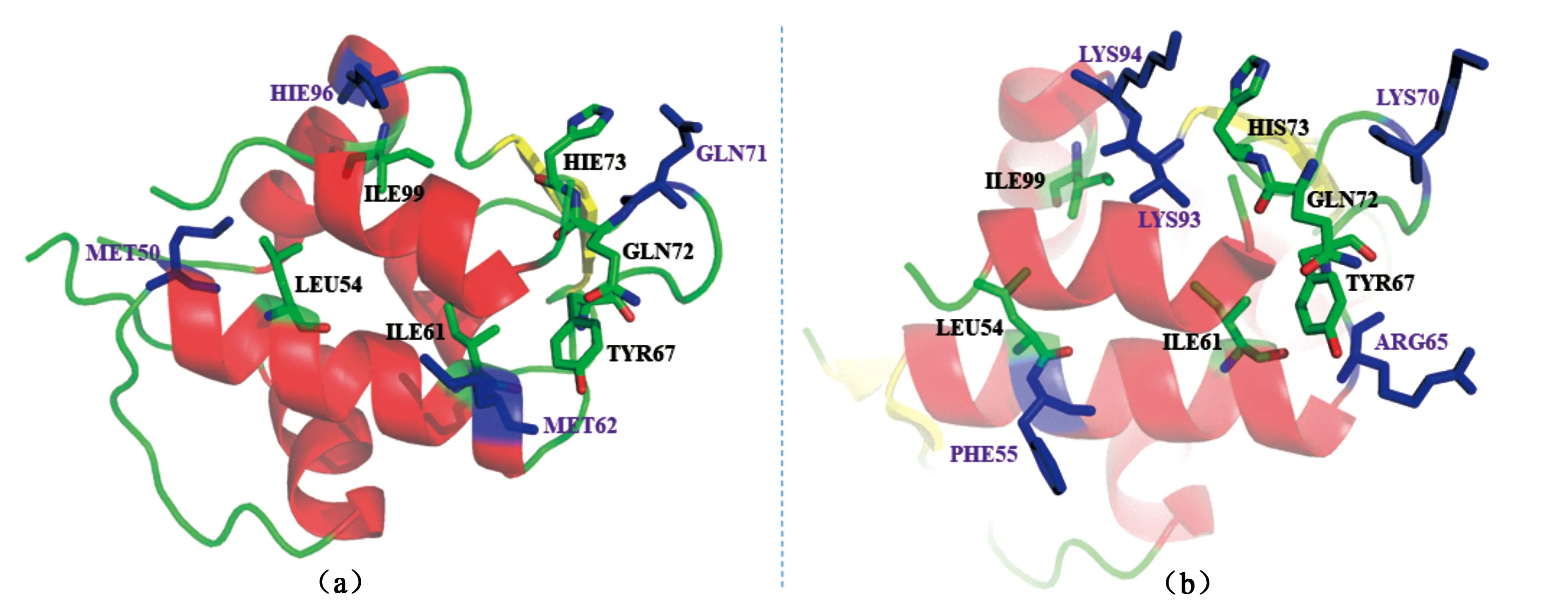
图3 MDM2体系关键残基相对位置图.(a)为p53- MDM2体系;(b)为pDIQ-MDM2;蓝色棍棒模型为两体系间差异残基.
2.6小结蛋白-配体,蛋白-蛋白是一种由潜在的物理的化学相互作用决定的特定生物识别过程,它几乎发生在整个生理过程中.对这些相互作用的研究最直接的实用价值就是促进药物的研发,所以关于它们的作用机理研究一直都层出不穷.除本章上文介绍的研究热门的蛋白质外,研究者对于其它的重要蛋白质的研究也取得一些突破性的进展.Zou等人[89]用IE方法计算了多肽构象熵的贡献,揭示了O4分子对人胰岛淀粉样多肽(Human Islet Amyloid Polypeptide, hIAPP)详细破坏机制:无论是hIAPP的五聚体还是十聚体,O4分子都是通过直接与残基22NFGAI26结合从而破坏了肽间的局部β片层.另外,相互作用熵理论除在研究蛋白-配体的相互作用机制上大放异彩之外,对于药物的虚拟筛选也能别开蹊径.2017年Li等人[90]使用相互作用熵从PubChem数据库中筛选出了AD的两种潜在药物:CID 16040294和CID 9998128.一些研究者也对相互作用熵的精确性进行了系统地评估:Biggin课题组对BRDs系列蛋白的复合物和抑制剂为Bromosporine的复合物进行了测试,发现当使用MM/PBSA计算蛋白-配体的结合自由能时,引入相互作用熵会极大的提高计算精确度,但是几乎不花费额外的计算成本[11].上述工作都表明了相互作用熵在计算蛋白-配体,蛋白-蛋白的结合自由能时具有相当好的准确性,无论是在计算精确度还是计算成本上都得到很好的改良.相互作用熵的引入不仅使得对蛋白质-配体的相互作用机制有了新的认识[88],也对蛋白-蛋白间相互作用能的计算提供了创新性的突破[91, 92].但是就总体工作而言,关于蛋白-蛋白相互作用的研究依旧是相对较少,这可能是因为蛋白-蛋白的相互作用的生物功能界面过大导致研究困难.我们也期待着未来对蛋白-蛋白相互作用有着更多的研究工作.
3 结 语
相互作用熵的引入极大提高了MM/PB(GB)SA方法预测生物大分子结合自由能的准确性,同时在计算速度上也实现了很大的提升.目前,相互作用熵方法结合MM/PB(GB)SA方法进行的结合自由能计算已经成功地应用于分子的相互作用机制研究和药物设计的许多方面.但是随着应用的增多,研究者也发现在目前的方法中仍旧存在一些不足之处.
首先是计算速度问题,相互作用熵必须结合MM/PBSA方法才能进行结合自由能的计算.而其中求解PB方程一直以来是一项计算量巨大工作,相较而言,计算相互作用熵的成本几乎可以被忽略.因此,在计算结合自由能时,所有的计算成本几乎归因于焓变的计算上.针对这一缺陷,目前有两种较为可行方案.
1) 软加速方法.就是在现有的计算水平上,使用多核CPU并行手段,或者是通过GPU求解PB方程.
2) 使用GB模型来代替PB模型计算极性溶剂化能.事实上,GB模型在计算速度方面的表现在近些年来得到了广泛的关注,但同时提高GB计算精度也是一项重大的挑战.对此,研究者提出了一系列的GB模型,如GBHCT[93, 94]、GBOBC[95]以及GBGBn[96-98]用以改善GB模型的计算精确度,Hou等人[61]也对这些模型进行了一些测试,在此不再进行赘述.
另一个广泛关注的问题是计算的准确性问题.结合自由能在计算精度方面的困难一直都是研究者渴望克服的难题.针对这一难题,有如下3个建议.
1) 改善MD模拟.通过三个方面可以进行相对应的调整.其一,调整MD模拟的时间尺度.Wang等人统计自2011年关于MM/PB(GB)SA的应用,总结发现MD模拟时间严重影响的MM/PB(GB)SA所计算的精确度.[99]其二,选择合适的力场.力场的改进也一直是研究的热点.AMBER自发展以来,一直都致力于其力场开发,且其力场也越来越具有针对性.例如针对蛋白质的protein.ff系列、针对DNA的DNA.bsc1和DNA.OL15、针对RNA的RNA.OL3和RNA.ROC以及针对水分子的water系列.同时也可以采用极化力场进行MD模拟.其三,选择显示溶剂模型进行MD模拟.研究表明显示溶剂水模型的MD模拟对于MM/PB(GB)SA是必不可少的[48].
2) 计算轨迹的选择.现在主流的计算是采用一条MD模拟轨迹,然后将MD轨迹分成复合物,受体和配体的轨迹用于MM/PB(SA)计算,即1A-MM/PB(GB)SA方法.Samuel等人提出了将复合物、受体以及配体分别进行MD模拟以得到三条互补干扰的轨迹,该方法被称为为3A-MM/PB(GB)SA.[100]关于这两种方法的优缺点Wang等人[99]已经进行了总结,对此也不再赘述.事实上,计算资源较为充足的情况,对多次1A-MM/PB(GB)SA求平均是一种可信度高的选择[69, 78, 101-103].
3) 内部参数的调整.可以根据不同的生物分子或者根据不同的残基类型采用不同的介电常数.该思想在MM/PBSA或者MM/GPBSA方法中都适用.另外,改变PB半径也可作为一种提高结合自由能精确度的尝试[104, 105].
相互作用熵方法除在计算结合自由能上有着优秀的表现外,将其运用在热点残基的预测上也是另辟蹊径.热点氨基酸预测中增加了熵变的贡献极大提高了预测的可信度,另外,相互作用熵的计算速度的优势也依旧被保留了下来.同时,将热点残基总能量求和用以计算整个体系的结合自由能,也为研究者计算生物大分子相互作用的结合自由能提供了新的可靠途径.该理论从提出到现在的四年里,因其理论严谨、计算成本低而被广泛用于研究蛋白-蛋白、蛋白-配体等生物大分子结合自由能计算中.但是关于其在另外一些研究热门的生物大分子体系上,像DNA-蛋白、RNA-蛋白、凝集素-糖等复合物上鲜有报道,相互作用熵理论也被期待着能在这些体系的研究上取得一些新的进展.
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
中国音乐学(2022年1期)2022-05-05
油气·石油与天然气科学(2021年9期)2021-10-10
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中学生物学(2019年1期)2019-01-13
教学与管理(理论版)(2017年7期)2017-08-11
浙江大学学报(理学版)(2016年6期)2016-12-15
天津科技大学学报(2015年2期)2015-08-09
中国药业(2014年17期)2014-05-26
