
白光LED用Mn4+激活红光荧光粉中锰离子价态表征研究进展
2020-10-13 13:46王兆武姬海鹏易莎莎侯星慧陈德良解荣军
发光学报 2020年10期
王兆武,姬海鹏*,徐 坚,易莎莎,侯星慧,陈德良,3*,解荣军
(1.郑州大学 材料科学与工程学院,河南 郑州 450001; 2.河南理工大学 物理与电子信息学院,河南 焦作 454000;3.东莞理工学院 材料科学与工程学院,广东 东莞 523808; 4.厦门大学 材料学院,福建 厦门 361000)
1 引 言
作为一种高效半导体光源,白光发光二极管(Light-emitting diodes,LEDs)正迅速取代荧光灯和白炽灯。在实现白光的多种途径中,蓝光LED复合Y3Al5O12∶Ce3+黄光荧光粉的方法,具有结构简单、高效稳定的优点而成为白光LED主流商业方案[1]。为了实现暖白光发光和提高显色指数,需要在上述体系中再加入红光荧光粉[2-3]。因此,蓝光LED泵浦新型红光荧光粉的研发具有重要价值。
当前成功商业化应用的红光荧光粉主要有Eu2+激活氮化物和Mn4+激活氟化物两大类。前者包括(Ba,Sr)2Si5N8∶Eu2+(λem=590~625 nm, 半高宽2 050~2 600 cm-1)[2]和(Ca,Sr)AlSiN3∶Eu2+(λem=610~660 nm,半高宽2 100~2 500 cm-1)[4]。此外,SrLiAl3N4∶Eu2+(λem=650 nm, 半高宽1 180 cm-1)[5]和Sr[Mg3SiN4]∶Eu2+(λem=615 nm,半高宽1 170 cm-1)[6]作为窄带红光荧光粉也引起了研究者们极大的兴趣。后者主要包括以K2SiF6∶Mn4+为代表的A2MF6∶Mn4+(A= Li+/K+/Na+/Rb+/Cs+;M=Si4+/Ge4+/Ti4+/Zr4+/Sn4+)氟化物[7]。K2SiF6∶Mn4+类氟化物荧光粉表现出来自于ν6/ν4/ν3振动模式、以630 nm为主峰的多个Stokes和反Stokes发射峰[8],每个发光峰都很窄(半高宽<5 nm),应用于液晶显示白光背光源技术时可实现广色域显示[9-10],当前市场应用份额逐年增加。但其缺点是易潮解、制备过程中需使用强腐蚀性氢氟酸等。为解决该问题,近年来也有大量Mn4+激活氧氟化物[11]和Mn4+激活氧化物[12]的研究。
K2SiF6∶Mn4+荧光粉潮解劣化时发生量子效率骤减的主要原因之一是Mn4+价态的变化,如潮解形成含Mn3+的KMnF4·H2O和K2MnF5·H2O[13],而在双85高温高湿环境下(85 ℃和85%相对湿度),荧光粉表面的[MnF6]2-容易快速水解为MnO2而变黑[14]。在研发新型Mn4+激活氧氟化物和氧化物荧光粉的过程中,锰离子的多价态共存问题也成为研发新型高效Mn4+激活红光荧光粉的困扰。然而,因为Mn4+离子d轨道电子波函数比稀土离子f轨道电子波函数更加扩展,更容易产生交互作用,因此Mn4+激活荧光粉中锰离子最佳掺杂浓度比Eu2+激活荧光粉中稀土离子的最佳掺杂浓度更低[15];在掺杂量非常少的情况下,准确定性甚至定量确定多种价态Mn离子的存在变得非常困难。目前很多相关研究论文对所制备荧光粉中锰离子价态未进行表征或采用的表征手段难以定性或定量确定多种共存价态锰离子的相对含量。针对该现状,本文对Mn4+激活红光荧光粉中锰离子价态的表征研究进行综述,含漫反射光谱、荧光光谱、X射线光电子能谱、电子顺磁共振谱、阴极射线发光谱、X射线精细吸收谱、变温磁化率谱,并对其结果可靠性和测试便易性进行对比评述。最后总结影响锰离子价态的因素并对Mn4+激活荧光粉中Mn4+价态调控进行展望。
2 锰离子的价态及常用锰源
锰属于过渡金属元素,电子构型为[Ar]3d54s2,具有未充满的d轨道。可依次失去外层轨道电子而呈现+7、+5、+4、+3、+2等多种氧化态。
文献中报道K2MnF6、MnCO3、MnO2、Mn(NO3)2或Mn(CH3COO)2等可作为Mn4+激活红光荧光粉的锰源。绝大多数Mn4+激活氟化物、氧氟化物荧光粉论文使用K2MnF6作为锰源,而绝大多数Mn4+激活氧化物荧光粉论文使用MnCO3为锰源。K2MnF6是一种极易水解劣化的锰源,需现用现制,其在后续Mn4+激活红光荧光粉制备以及在热和潮湿环境下的劣化以及锰价态演变可参见Smet等的研究[13,16]。本文侧重于综述Mn4+激活氧化物红光荧光粉制备所用锰源以及其价态表征手段。
(1)MnCO3是绝大多数Mn4+激活氧化物红光荧光粉制备时所用锰源。Chen等[17]报道MnCO3在加热过程的物相演变为:
也有文献报道了稍有差异的物相演变过程。Xu等[18]报道了MnCO3在加热过程中的热重(TG)与差热(DTA)曲线(图1(a))及其在不同温度保温1 h后的XRD(图1(b))。从室温加热到1 000 ℃过程中,MnCO3出现3个阶段质量损失:室温到450 ℃,出现约30%质量损失;450~900 ℃,出现约5%质量损失;900~930 ℃,出现约2%质量损失。从图1(b)可以看出,加热到450 ℃时,MnCO3氧化为MnO2,加热到600~750 ℃之间出现Mn2O3,进一步提高温度时出现Mn3O4。因此,MnCO3在空气中逐渐升温过程中先氧化后还原,可表述为:MnCO3-CO2+1/2O2→MnO2; 2MnO2-1/2O2→Mn2O3; 3Mn2O3-1/2O2→2Mn3O4。尽管MnCO3自身在加热过程中会依次呈现不同价态,但在制备荧光粉时锰离子掺杂进入基质化合物晶格,其最终所呈现的价态不仅受温度影响,更受到所掺杂格位晶体结构[19]、气氛、电荷补偿剂等因素影响,因此使用MnCO3作为锰源经高温处理后可得到Mn4+掺杂荧光粉。
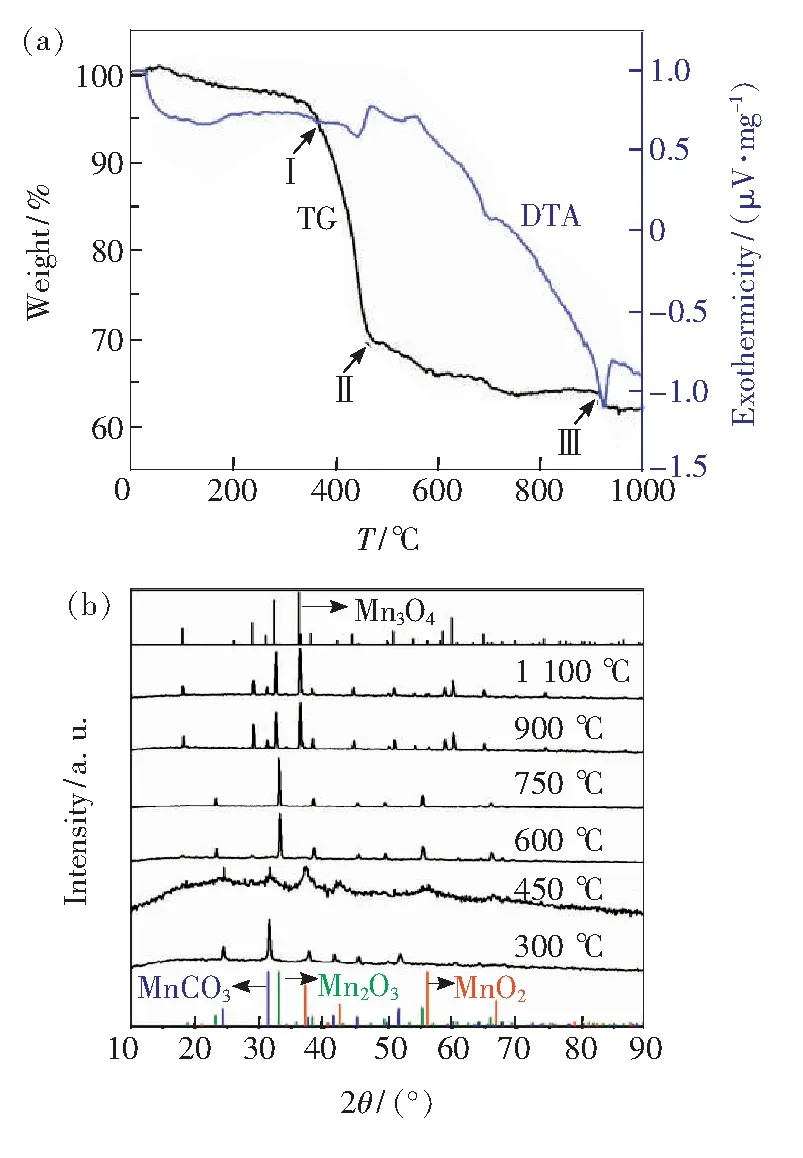
图1 (a)MnCO3在室温至1 000 ℃范围内空气中以10 ℃/min加热所得TG/DTA曲线;(b)MnCO3在不同温度下加热1 h后的XRD图谱及MnCO3(PDF#11-3970)、Mn2O3(PDF#41-1442)、MnO2(PDF#30-0820)和Mn3O4(PDF#24-0734)的标准卡片[18]。
(2)其他一些文献报道了使用MnO2作为锰源。Weng等[20]用Li2CO3、MgO、TiO2为原料,MnO2为锰源,经1 100 ℃保温4 h制得Li2MgTi3O8∶Mn4+荧光粉。Amarasinghe等[21]用NaCO3、MgO、WO3为原料,MnO2为锰源,经675 ℃保温12 h制备了从4T2g能级跃迁回4A2g基态能级而呈宽带发光的Na4Mg(WO4)3∶Mn4+荧光粉。Peng等[22]使用MgO、TiO2、GeO2为原料,MnO2为锰源,经1 400 ℃保温6 h制得Mg2Ti1-xGexO4∶Mn4+荧光粉。Naresh等[23]采用K2CO3、Ga2O3、NH4H2PO4为原料,MnO2为锰源,经950 ℃保温10 h制得KGaP2O7∶Mn4+深红光荧光粉。使用MnO2为锰源的文献数量比以MnCO3为锰源的文献数量少很多,原因可能是MnO2的化学性质相对不活泼、原料颗粒较大、不易得到高纯原料或其他未知原因。
(3)少量文献报道了使用Mn(NO3)2或Mn-(CH3COO)2作为锰源。Ali等[24]以MgO、GeO2和MgF2为原料,使用稀释的Mn(NO3)2·4H2O溶液为锰源,以便精确控制所制备Mg28Ge7.5O38F10∶Mn4+红光荧光粉中锰离子的添加量,有效解决了所需掺杂量因量少而称量不准确的问题。Ji等[25]用MgCl2·6H2O、Al(NO3)3·9H2O和氨水为原料,稀释的Mn(NO3)2溶液为锰源,经共沉淀法得到前驱体并再经热处理制得主峰为651 nm的MgAl2O4∶Mn4+红光荧光粉。Zhang等[26]用Mg(CH3COO)2·4H2O、LiCO3、TiO2、柠檬酸为原料,Mn(CH3COO)2·4H2O为锰源,经溶胶-凝胶法制得Li2MgTi3O18∶Mn4+深红光发光荧光粉。因此,当合成少量样品欲精确控制锰源的称量,或者采用共沉淀法、溶胶-凝胶法等湿化学方法合成Mn4+掺杂荧光粉时,可用稀释的硝酸锰或醋酸锰溶液作为锰源。
3 锰激活荧光粉中锰离子价态表征
相关文献分别采取了一种或两种手段来表征所合成Mn4+激活红光荧光粉中锰离子的价态。本文将其总结归纳为如下7种手段,即漫反射光谱(及颜色)、荧光光谱、X射线光电子能谱、电子顺磁共振谱、阴极射线发光光谱、X射线吸收精细结构谱、变温磁化率谱。
3.1 漫反射光谱和颜色
当荧光粉中所掺杂锰离子价态不同时,因其所表现的能级跃迁吸收不一样而在紫外-可见-红外光区表现出不同的漫反射光谱。锰离子最外层为d轨道,是非球形对称轨道,在晶体场作用下d轨道发生能级劈裂,形成不同的能级;电子在不同能级间跃迁,跃迁能在1~4 eV之间,对应吸收一定可见光区范围的光,使物质呈色(所呈颜色为其所吸收光的补色,在太阳光或其他光源下所呈颜色根据锰元素价态不同而不同)。因此,可通过对比所合成荧光粉颜色及漫反射光谱来初步分析所含锰离子的价态。新制备Mn4+激活氟化物荧光粉呈橙黄色或粉红色,其中锰离子主要以+4价存在,其漫反射光谱主要包括对应于Mn4+离子d-d跃迁的吸收。Du等[27]通过两步共沉淀法制备了K2TiF6∶Mn4+荧光粉,图2(a)为他们测得的室温漫反射光谱和激发光谱。漫反射光谱中位于360 nm和460 nm的两处吸收分别可归属于Mn4+离子4A2g→4T1g和4A2g→4T2g跃迁,这与其激发光谱的特征峰相对应。而在Mn4+激活氧化物荧光粉中,除观测到对应于Mn4+离子d-d跃迁的吸收外,还观测到O2-→Mn4+电荷迁移跃迁吸收。退火处理是导致荧光粉中锰离子价态改变的一种方法。Zhang等[28-29]通过真空烧结并在氧气中不同温度下退火制备了Lu3Al5O12∶Mn透明荧光陶瓷,研究了退火条件对锰离子价态的影响。图2(b)为真空烧结所制备Lu3Al5O12∶Mn陶瓷和其在O2中不同温度退火处理后的直线透过率(插图为420~520 nm范围的放大图及该系列荧光陶瓷经退火处理后的实物照片)。除450 nm附近、对应于4A2g→4T2g跃迁的吸收外,还观测到200~350 nm范围的吸收,其为4A2g→4T1g跃迁和O2-→Mn4+电荷迁移跃迁的叠加。通过在氧气气氛中进行退火处理,显著改变了Lu3Al5O12∶Mn陶瓷中锰离子的赋存价态。随着退火温度升高,样品在紫外区的吸收发生了红移,且吸收强度随热处理温度升高而增加。从插图所示蓝光区(420~520 nm)的局域吸收带可以看出,当退火温度升高到1 300 ℃,该区域开始出现吸收带,并随温度继续升高到1 600 ℃而达到最强。上述在紫外和蓝光区的新的吸收带归属于Mn4+的电荷迁移跃迁和d-d跃迁。因此,退火后锰离子价态逐渐变为Mn4+为主,热处理温度是将Mn3+/2+氧化为Mn4+的重要影响因素。真空烧结所得陶瓷样品为无色透明,在O2中1 200~1 600 ℃退火后,颜色不断变化:1 200,1 300 ℃热处理样品为浅粉色,≥1 400 ℃热处理样品为橙色。样品颜色变化归因于Mn离子价态及相应吸收特性的变化。
Liao等[30]以CaCO3、ZnO、Ga2O3为原料,MnO2为锰源,经1 230 ℃保温6 h制备了Ca14Zn6Ga10O35∶Mn深红光发光荧光粉。在图2(c)所示的漫反射光谱中,除观察到上述对应于电荷迁移带跃迁和Mn4+离子d-d跃迁的吸收外,还观测到580~800 nm范围宽的吸收。由于作者在该荧光粉中观察到主峰位于1 152 nm、来自于Mn5+离子的窄带发光,监测该发光所得激发光谱中观察到580~800 nm的宽激发带,因此作者认为漫反射光谱中580~800 nm的吸收来自于所制备Ca14Zn6Ga10O35∶Mn荧光粉中的Mn5+杂质离子,且Mn4+与Mn5+之间存在有效的能量转移。
由所测漫反射光谱数据换算得到的吸收光谱也能清晰地分辨所制备荧光粉中锰离子的价态。Xe灯照射是导致氟化物荧光粉中锰离子价态改变的一种因素。Hoshino等[31]通过共沉淀法合成了ZnGeF6·6H2O∶Mn4+红光荧光粉并研究了其在Xe灯照射下的劣化行为。图2(d)所示为所制备ZnGeF6·6H2O∶Mn4+红光荧光粉在Xe灯照射前/后的吸收光谱。Xe灯照射前,其在~460 nm和~360 nm处有两个吸收峰,分别对应于Mn4+离子4A2→4T2和4A2→4T1跃迁。Xe灯照射5 min后,显示出不同的吸收峰,在紫外区(<400 nm)的吸光度α急剧增加,可能是由于Mn3+或Mn5+吸收所致。此时荧光粉由浅柠檬黄变为浅粉黄色。由此可见,吸收光谱也可用于表征荧光粉中锰离子的价态。
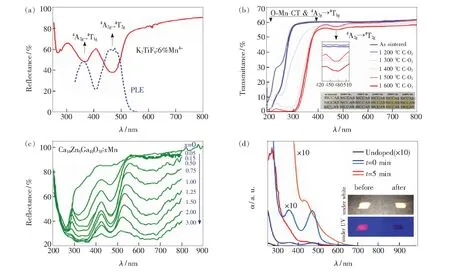
图2 (a)K2TiF6∶Mn4+荧光粉的漫反射光谱(虚线所示为其激发光谱)[27];(b)真空烧结所制备Lu3Al5O12∶Mn透明荧光陶瓷样品和在O2中不同温度下退火后的直线透过率(插图为420~520 nm范围的放大图及该系列荧光陶瓷经热处理后的实物照片)[29];(c)Ca14Zn6Ga10O35∶xMn荧光粉的漫反射光谱[30];(d)ZnGeF6·6H2O∶Mn4+荧光粉使用Xe灯照射5 min前后的室温吸收光谱(由漫反射数据换算所得),也测试了未掺杂的ZnGeF6·6H2O粉末样品[31]。
目前漫反射光谱的表征主要集中在短波长区域(200~800 nm)。随着中红外和远红外光区光电探测器技术的成熟化,越来越多的漫反射光谱仪可同时进行从紫外到中远红外光区的测试。Smet等[13]测试了更宽波长范围(200~1 400 nm)的漫反射光谱,以其为手段研究K2MnF6和K2SiF6∶Mn4+在潮湿环境下的水解变质行为。通过测试MnF2、MnF3、KMnF4·H2O/K2MnF6、K2MnF6、K2MnF6/K2MnF5·H2O/KHF2和老化的K2MnF6样品的漫反射图谱,研究了Mn3+形成对K2MnF6光吸收的影响[13]。图3(a)为其漫反射光谱,图3(b)为部分波长区域放大。圆括号代表该粉末样品由多种晶相组成。样品老化条件为60 ℃/50%相对湿度。MnF2为Mn2+的参比物,在长波长到短波长区可以观察到一个光滑的宽谱,没有尖锐的吸收峰。MnF3为Mn3+的参比物,在432 nm和518 nm处存在两个尖锐的小峰,并在800 nm处存在一个宽峰。此外,在664 nm以左和413 nm以左观察到了漫反射率下降,在漫反射光谱中形成阶梯状特征。在(KMnF4·H2O/K2MnF6)的漫反射光谱中观察到了相似特征,证明在水合物中存在Mn3+。(K2MnF6/K2MnF5·H2O/KHF2)的漫反射光谱中在800 nm处也可以观察到归属于Mn3+的宽谱。老化的K2MnF6同样观察到了相似的特征。即使是纯的K2MnF6(XRD显示无杂相),也观察到除了450 nm处K2MnF6的Mn4+的吸收峰外在长波长部分的伴峰(归属于Mn3+),说明即使是纯的K2MnF6仍存在有相当一部分的Mn3+。
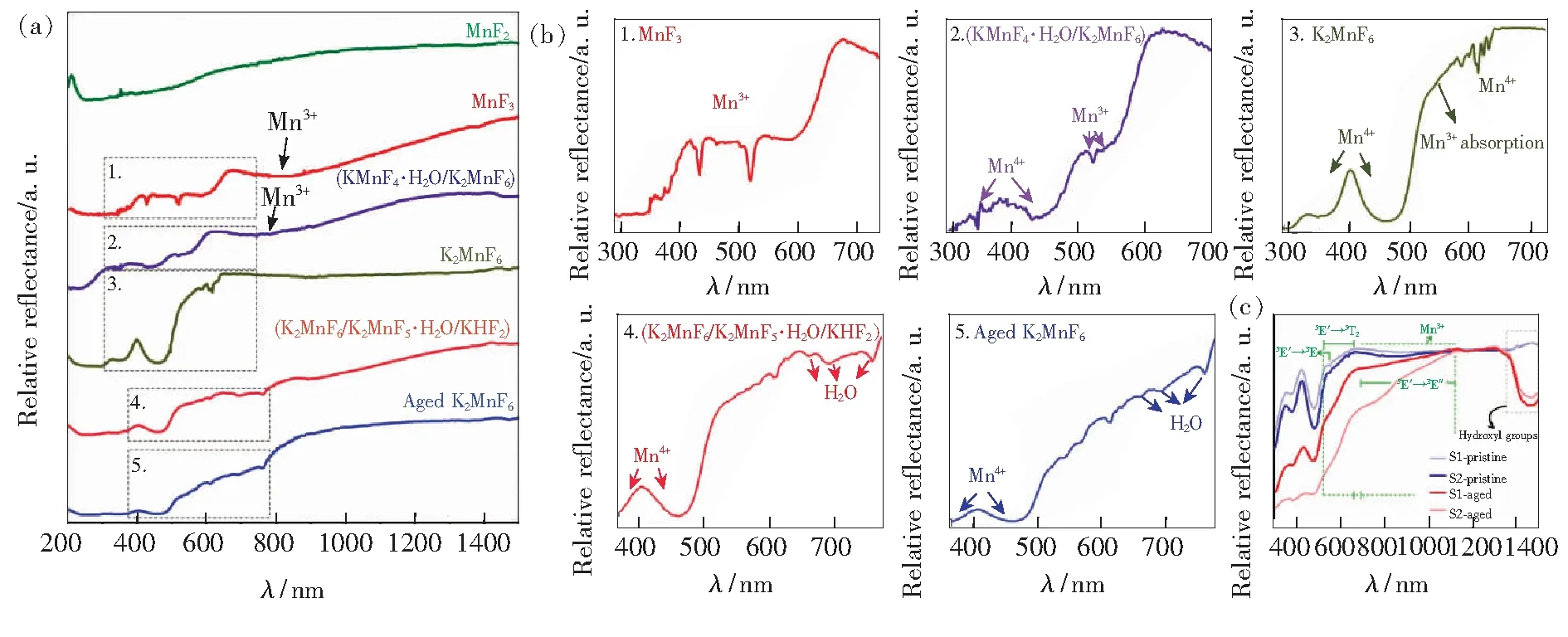
图3 (a)MnF2、MnF3、K2MnF6等氟化物的漫反射光谱;(b)图(a)的漫反射光谱中部分波长区间的放大图[13] ;(c)含有KHF4杂质的K2SiF6∶Mn4+(样品S1)在老化前后(70 ℃/80%相对湿度环境中放置24 h)的漫反射光谱,样品S2中还包含了K2MnF6杂质[32]。
此外,该课题组还研究了K2SiF6∶Mn4+合成和老化过程中杂质形成的起源及造成的后果[32]。KHF2杂相和Mn3+杂质离子都影响了荧光粉的性能。Mn3+主要影响光吸收行为。KHF2杂相不仅影响光吸收,而且还影响荧光粉的化学稳定性。图3(c)显示纯K2SiF6∶Mn4+荧光粉在70 ℃/80%相对湿度条件下老化48 h,与含有KHF2杂相样品仅老化20 h的漫反射光谱的对比。纯相样品在345 nm和450 nm处的两个吸收峰分别对应于Mn4+的4A2g→4T1g和4A2g→4T2g跃迁,无其他吸收峰出现。而含有杂相的样品在450 nm处出现一个伴随吸收峰。不管是原始的还是老化后的样品,在500~620 nm之间都存在寄生吸收,而且在800 nm处存在宽的吸收峰,这都归因于Mn3+。图3(c)中的样品显示出与KMnF4·H2O、MnF3类似的吸收。MnF3中518 nm处的吸收峰归属于Mn3+离子的5E′→3E自旋允许跃迁。在氟化物中5E′→5T2的跃迁为介于410~620 nm之间宽而强的吸收带。图3(c)中Mn3+的5E′→5T2吸收带为Mn4+离子4A2g→4T2g吸收带低能量边的肩峰。Mn3+离子的这一自旋允许吸收带具有高的振子强度,可以用于检测制备的氟化物荧光粉中的Mn3+。Mn3+杂质离子会导致K2SiF6∶Mn4+的寄生吸收,造成量子效率降低。
由上述实验结果可以看出,当荧光粉中所含锰离子的价态不同时,对光的吸收不同而表现出迥异的漫反射光谱(可以漫反射率、吸收率或透过率对不同形态样品进行评价)。Mn4+在280~320 nm的吸收带属于O2-→Mn4+电荷迁移带和4A2g→4T1g自旋允许跃迁,350~520 nm范围的吸收分别来自4A2g→2T2g自旋禁戒跃迁和4A2g→4T2g自旋允许跃迁;Mn2+在400~500 nm之间产生来自于6A1→4T2和6A1→4A1跃迁的吸收带。Mn3+和Mn5+也会导致紫外区(<400 nm)的吸光度增大。荧光粉中锰离子发生价态改变的因素有水解、Xe灯照射、受热和退火处理等。锰离子的价态变化会使荧光粉呈现出不同的颜色。一般地,所制备荧光粉中Mn4+为主时,体色呈橙黄色或粉红色;Mn2+为主时,体色呈白色;Mn3+为主时,可能呈灰色。
3.2 荧光光谱
荧光粉的发光特性可由激发与发射光谱表征。在可见光区能观测到的荧光光谱主要来自形成八面体配位的Mn4+及形成四面体或八面体配位的Mn2+。由于Mn4+和Mn2+的能级跃迁特征差异明显,因此其表现出迥异的光致激发与发射光谱[7]。
图4(a)所示为ZnGeF6·6H2O∶Mn4+红光荧光粉的室温发射和激发光谱[31]。Mn4+的有效离子半径r=0.053 nm,可以替代[GeF6]2-中的Ge4+(r=0.053 nm),而Mn2+(r=0.067 nm)不能。ZnGeF6·6H2O∶Mn4+红光荧光粉表现出Mn4+激活荧光粉的典型的尖锐线状红光发射,归属于Mn4+的2Eg→4A2g跃迁。PLE谱在470 nm和370 nm处有两个宽的激发带,分别隶属于Mn4+离子的4A2g→4T2g和4A2g→4T1g跃迁。
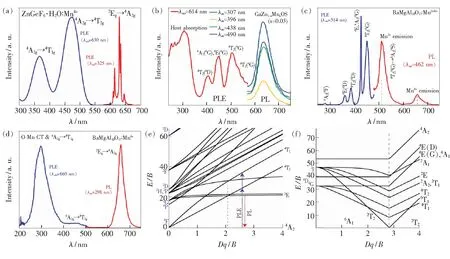
图4 (a)ZnGeF6·6H2O∶Mn4+的室温发射(PL)光谱和激发(PLE)光谱[31];(b)CaZn1-xMnxOS(x=0.03)的PL/PLE光谱[34];(c)BaMgAl10O17∶Mn2+/4+的PL/PLE光谱[33];(d)BaMgAl10O17∶Mn4+的PL/PLE光谱[33];d3离子(e)和d5离子(f)的Tanabe-Sugano图。
Mn2+离子通常显示出更宽的对应于4T1→6A1跃迁的发射带。Mn2+的发射能量受到晶体场强度的强烈影响,可在绿光至深红区内变化(500~750 nm)。Mn2+形成四配位时受弱晶体场作用而表现绿光发射,形成八配位时受强晶体场作用而表现橙色或红光发光。图4(b)所示为CaZnOS∶Mn2+红光荧光粉的激发与发射光谱[31]。CaZnOS∶Mn2+表现出主峰波长为614 nm处、对称的红光发射带(半高宽为50 nm),归属于Mn2+离子4T1(4G)→6A1(6S)跃迁。在CaZnOS中Mn2+取代Zn2+形成[MnS3O]四面体,通常Mn2+在四面体配位时发射绿光,而在CaZnOS基质中却发出红光,这是由于Mn2+处于畸变四面体配位时,二能级系统进一步劈裂为三能级,使得第一激发态向低能量区移动所致。CaZnOS∶Mn2+的激发光谱涵盖230~550 nm区域,其中短波处的强激发带(<350 nm)源自晶格基质,而350~550 nm之间的激发带分别归属于基态6A1(6S)到4T2(4D)、[4A1(4G)、4E(4G)]、4T2(4G)、4T1(4G)的跃迁。由于Mn2+的d-d跃迁是自旋和宇称禁戒的,因此直接激发Mn2+的激发态能级,强度非常弱;而CaZnOS∶Mn2+的激发带却相当强,是因为强共价的[MnS3O]混合配位使得Mn2+的d-d跃迁解除了自旋和宇称禁戒。
当同一基质中同时含有Mn4+和Mn2+时,其表现出迥异的荧光光谱。Xu等[33]在空气气氛下经高温固相法合成了BaMgAl10O17∶Mn0.01荧光粉,其荧光光谱示于图4(c)、(d)。图4(c)所示荧光粉中Mn主要以+2价形式存在,少量以+4价形式存在。在462 nm蓝光激发下,荧光粉表现出峰值为514 nm的典型Mn2+绿光发射带;而在298 nm激发下,在660 nm处存在一个比较弱的Mn4+特征发射峰。图4(d)所示荧光粉中Mn主要以+4价形式存在,其激发和发射光谱与Mn4+激活铝酸盐荧光粉吻合。本课题组[25]曾报道共沉淀法制备MgAl2O4∶Mn荧光粉中因热处理制度不同而引起的锰离子选择性格位占据现象,即不同热处理制度时,Mn可能以+4价形式取代MgAl2O4中六配位Al3+格位,也可能以+2价形式取代四配位Mg2+格位,因此表现出迥异荧光光谱。
Mn4+与Mn2+的荧光特征迥异的现象可由d3和d5离子的Tanabe-Sugano图进行解释(示于图4(e)、(f)。Mn4+的发射峰为窄带发射(620~750 nm),归属于2Eg→4A2g禁戒跃迁,Mn4+激活的氟化物荧光粉为典型的锐线红光发射带,而Mn4+激活氧化物荧光粉则多为不对称的宽峰发射。Mn2+的发射带范围较广(500~750 nm),可在绿光至深红区变化,发射带较对称,来自于4T1(4G)→6A1跃迁,发射峰受晶体场强烈影响。Mn4+的PLE谱主要由4A2g→4T1g和4A2g→4T2g激发带组成,而Mn2+的PLE谱则由6A1→4E(4D)、6A1→4T2(D)、6A1→[4A1(G),4E(4G)]和6A1→4T1(G)组成。
3.3 X射线光电子能谱
X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)利用高能X射线(光子能量1 000~1 500 eV)照射样品,使样品表面所含元素的内层电子或价电子受激从原子核心壳层逸出,基于所发射电子(称光电子)的动能进行元素检测。所发射光电子的动能由激发光子的能量和功函数决定。可测量光电子的能量,以光电子的动能/束缚能(Binding energy,Eb=hν(光能量)-Ek(动能)-w(功函数))为横坐标、相对强度(脉冲/s)为纵坐标做出光电子能谱图。光电子是由样品表面几个纳米范围内发射的,因此XPS是一种对表面敏感的技术。因原子周围电子处于不同价态时所拥有能量不同,受激时所需能量也就不同,电子跃迁时会有不同的现象发生而可判断其价态。Xu等[33]研究了α-Al2O3∶Mn荧光粉中锰离子价态及共掺电荷补偿剂Mg2+/Si4+对其发光性能的影响。图5(a)表明,所有荧光粉(单独Mn掺杂、Si/Mn共掺杂和Mg/Mn共掺杂)中Mn 2p3/2核心能级的XPS信号峰都出现在642.60 eV附近。Mn2+2p3/2(MnO)、Mn3+2p3/2(Mn2O3)和Mn4+2p3/2(MnO2)的峰值分别为641.0,641.9,642.6 eV,因此可推断α-Al2O3∶Mn中锰离子主要为+4价。然而,Si/Mn共掺样品的XPS图中在约647 eV处出现一个肩峰而其他两个样品中没有出现。肩峰可能是对应于MnO的卫星特征峰,因此Si4+共掺时诱导了Mn2+的出现。
Mn2+是除Mn4+外在可见光区有重要应用的锰离子。为区别两者,以下给出Mn2+激活荧光粉的XPS实例。Mn2+激活荧光粉常在还原气氛(H2、N2/H2或CO)下制备,但在某些基质中,即使是在强还原气氛下合成,Mn的化合价也很难保持在Mn2+。单价Mn4+或Mn2+发光可以通过掺杂电荷补偿剂(Mg2+、Ca2+、Ln3+、Bi3+)获得。Wei等[35]使用不同Mn源(MnCO3/MnO2/Mn2O3/KMnO4)在空气下合成了Na2ZnSiO4∶Mn2+荧光粉并研究锰离子的自还原机制。利用XPS和EPR对样品进行了检测。由图5(b)可以看出,所有样品都在约642.1 eV和655.0 eV存在两个峰,其分别对应Mn2+的2p3/2和2p1/2能级,这表明Mn7+、Mn4+、Mn3+、Mn2+在系统中确实发生了自还原过程。Xu等[18]研究了缺陷诱导Mn2+发光和不同价态锰源(MnCO3和MnO2)对Mn2+发光的影响机制以及Mn2+的自还原过程。图5(c)为其所合成Li2CdSiO4∶Mn2+荧光粉Mn 2p轨道高分辨XPS谱。峰值为641.1 eV和652.4 eV的两个XPS峰与Mn 2p3/2和Mn 2p1/2的键能较好吻合,说明锰离子的主要存在形式为Mn2+。
前述漫反射光谱和荧光光谱都难以定量描述荧光粉中多种锰离子之间的定量关系,但据文献报道,可依靠XPS技术对所制备荧光粉中同时存在的Mn4+及Mn2+进行定量表征。Dong等[36]使用高温固相法合成了Gd3Ga5-x-δAlx-y+δO12∶yMn荧光粉,并通过阳离子替位策略调控荧光粉中锰离子的价态,即通过调控Al3+/Ga3+的固溶浓度来调控Mn4+/Mn2+离子的比例。图5(d)所示为Gd3Ga5-x-δAlx-y+δO12∶yMn荧光粉中Mn离子的高分辨XPS图谱。其中,结合能为659.5,660.1,643.7,646.7 eV的XPS峰分别可归属于Mn2+2p1/2、Mn4+2p1/2、Mn2+2p3/2和Mn4+2p3/2,因此可证实合成的样品中存在Mn2+和Mn4+,且Mn4+/Mn2+的含量比例约为0.42。
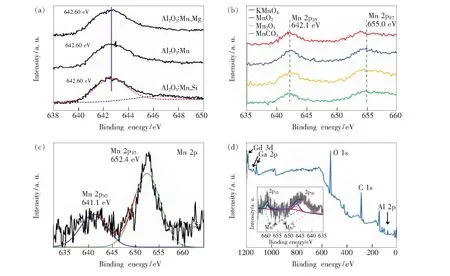
图5 (a)Al2O3∶Mn、Al2O3∶Mn,Mg和Al2O3∶Mn,Si样品的XPS谱[33];(b)不同Mn源合成的Na2ZnSiO4∶0.015Mn2+的XPS谱[35];(c)Li2CdSiO4∶0.02Mn荧光粉中Mn 2p轨道高分辨XPS谱[18];(d)Gd3Ga2-δAl2.998+δO12∶0.012Mn荧光粉的XPS谱(插图所示为Mn 2p轨道高分辨XPS谱)[36]。
由上述结果可以看出,Mn 2p核心能级的XPS图谱由Mn的2p1/2和2p3/2组成。通过所测XPS谱信号结合能与相应价态Mn 2p核心能级的键能比对可判定荧光粉中锰离子价态。相比于漫反射光谱和荧光光谱而言,XPS技术有望定量给出不同价态锰离子相对含量,但由于荧光粉中锰元素掺杂量很低(一般为0.05%~0.5%),XPS谱中对应于Mn 2p轨道高分辨谱呈现较大噪音,利用其定量多种价态锰离子的相对含量的可靠性存疑。
3.4 电子顺磁共振谱
电子顺磁共振(Electron paramagnetic resonance,EPR)是由未配对电子的磁矩发源的一种磁共振技术。原子核外层存在自旋磁矩不为0的电子,此时给以磁场,外层自旋磁矩不为0的电子的自旋能级发生分裂(称塞曼分裂);在外磁场垂直方向施加不同频率ν的微波,当满足共振条件hν=gβH时(h为普朗克常量,ν为微波频率,H为磁场强度,β为玻尔磁子,g为g因子),电子在分裂的能级之间发射跃迁,产生EPR信号(称为扫场法)。对自由基而言,轨道磁矩几乎不起作用,总磁矩的绝大部分(99%以上)的贡献来自电子自旋,所以电子顺磁共振亦称“电子自旋共振”(Electron spin resonance,ESR)。早期研究认为共振跃迁过程只有电子自旋磁矩的贡献而采用ESR这个术语;其后发现仅用电子自旋跃迁无法完全解释相关实验结果,尤其是来自过渡金属离子的现象;也就是电子轨道磁矩对于跃迁也有贡献,所以逐渐用EPR取代ESR。
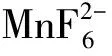
在相同测试条件下,随着Mn4+激活荧光粉中所含锰元素的浓度增大,EPR谱中这6个超精细结构也将愈加明显。Ye等[37]通过溶胶凝胶法合成Mg2TiO4∶Mn4+荧光粉并采用EPR研究掺杂Mn离子的价态。图6(b)所示为其室温EPR谱(扫描频率9.45 GHz),显示出6个超精细结构(150~210 mT),属于Mn4+的特征EPR信号;随着Mg2TiO4∶Mn4+中Mn4+掺杂浓度从nMn/(nMn+nTi)= 0.01%增大为0.25%,EPR信号强度明显增强。
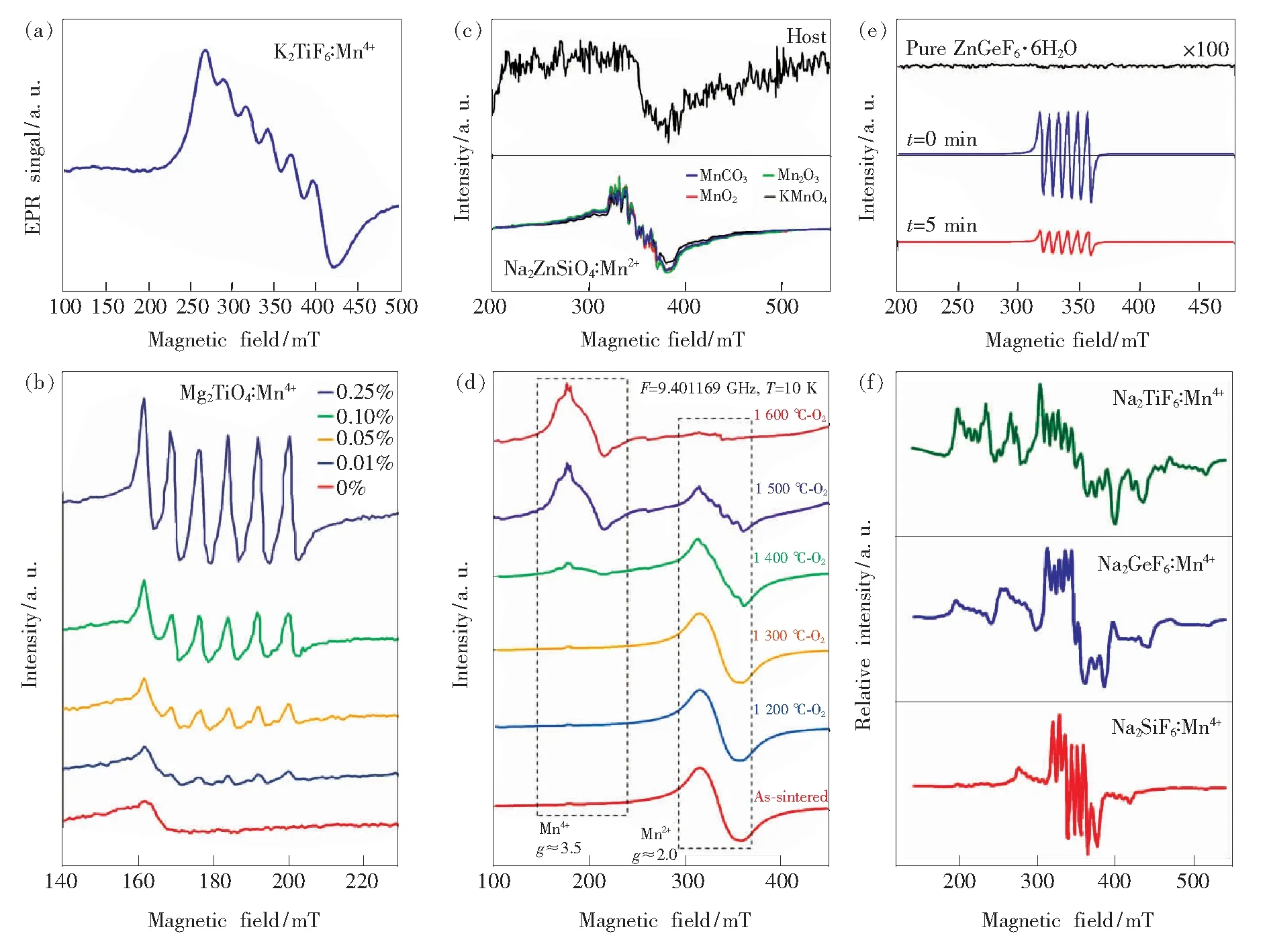
图6 (a)K2TiF6∶Mn4+荧光粉的EPR谱[27];(b)Mg2TiO4∶Mn4+荧光粉的EPR谱,nMn/(nMn+nTi)=0%,0.01%,0.05%,0.10%,0.25%[37];(c)由不同Mn源所合成的Na2ZnSiO4∶0.015Mn2+和其未掺杂基质的EPR谱[35];(d)真空烧结所得Lu3Al5O12∶Mn荧光陶瓷样品和在O2气氛中不同温度后期热处理后的EPR谱[29];(e)未掺杂ZnGeF6·6H2O基质以及ZnGeF6·6H2O∶Mn4+荧光粉在氙灯照射前后(t=0,5 min)的室温EPR谱[31] ;(f)Na2TiF6∶Mn4+、Na2TiF6∶Mn4+和Na2TiF6∶Mn4+的EPR谱[39]。
在Mn2+激活荧光粉中也观察到6个超精细结构的特征EPR谱。Wei等[35]用不同锰源(MnCO3,Mn2O3,MnO2,KMnO4)与原料Na2CO3、ZnO、SiO2混合,经高温固相法合成Na2ZnSiO4∶0.015Mn2+荧光粉,研究了合成过程中锰离子价态自还原现象。图6(c)所示为使用不同锰源合成的Na2ZnSiO4∶Mn2+及其基质的室温EPR谱。可以看出,Na2ZnSiO4基质的EPR谱中包含一个中心位于340 mT的信号较弱的宽带,可能来自于基质中的某固有缺陷;而Mn掺杂样品在300~400 mT范围可观察到相似的6个超精细结构EPR信号。该作者认为,EPR信号相同表明各荧光粉样品中Mn离子中的浓度和环境相似,这证明在制备过程中发生了高价Mn向Mn2+的自还原。此外,Singh等[38]认为在立方结构中Mn2+的EPR信号一般应出现在g≈2.0,且由于未配对电子与55Mn(核自旋量子数I=5/2)间相互作用而使EPR信号进一步劈裂为6个超精细结构;在低对称环境下,将出现对应于5/2↔3/2、3/2↔1/2、1/2↔-1/2、-1/2↔-3/2、-3/2↔-5/2跃迁的精细结构,但由于显著的各向异性,通常只观察到一个对应于1/2↔-1/2的伴随其6个超精细结构的宽信号峰,而对应于其他跃迁的EPR信号峰常无法分辨。而且,作为一种S态离子,Mn2+对EPR检测技术更加敏感,其信号更强[38]。
当荧光材料中Mn4+/Mn2+共存时,也有文献报道利用EPR来表征Mn4+/Mn2+的演变。Zhang等[29]使用EPR技术研究退火处理对Lu3Al5O12∶Mn中Mn价态影响。图6(d)为10 K条件下测得的不同热处理温度样品的EPR谱。样品在g≈2处呈现出宽而强的共振信号。该处的共振信号随着后期退火处理的温度升高而减弱,Mn离子的超精细结构逐渐出现,这些共振信号归属于Mn2+。结果表明,Mn2+含量随着后期热处理温度升高而减少。在1 400 ℃时,g≈3.5处出现的共振信号属于Mn4+。通过Mn2+和Mn4+共振信号的强度对比,可明确区分Mn2+和Mn4+在样品中的相对含量。温度升高至1 600 ℃时,Mn2+几乎全部氧化为Mn4+,此时发光强度明显提高。
此外,有文献报道认为Mn3+和Mn5+离子对EPR静默,不产生相关信号,但却可研究荧光粉中Mn4+离子向Mn3+/5+的转变[31]。图6(e)所示为室温下无掺杂ZnGeF6·6H2O和掺锰荧光粉ZnGeF6·6H2O∶Mn4+经Xe灯照射前后(t=0, 5 min)的EPR图谱[31]。ZnGeF6·6H2O的EPR谱中没有Mn相关信号,而ZnGeF6·6H2O∶Mn4+的EPR谱在~330 mT处出现6个特征精细结构,对应于Mn4+离子。当Xe灯照射后,ZnGeF6·6H2O∶Mn4+(t=5 min)中EPR信号强度大幅减弱,Mn4+的自旋密度大幅下降,作者认为这是源于Mn4+转变成了不产生EPR信号的Mn3+或Mn5+,即发生了光氧化Mn4+→Mn5+或歧化反应2Mn4+→Mn3++Mn5+[31]。
最后值得一提的是,Mn4+激活荧光粉的EPR谱可能会出现多个伴峰信号,该伴峰信号不应被解释为其他价态锰离子,而是来源于荧光粉中Mn4+微观配位八面体的畸变。Fang等[39]对比了Na2SiF6∶Mn4+、Na2GeF6∶Mn4+和Na2TiF6∶Mn4+荧光粉的EPR谱(示于图6(f))。所得EPR信号来自于顺磁性Mn4+离子。其中Na2SiF6∶Mn4+的EPR谱中包含4个信号(每个信号含有6个特征峰),分别位于磁场强度220,290,340,400 mT位置。最强EPR信号峰位于340 mT,对应于具有较高格位对称性的、孤立的Mn4+;剩余EPR信号峰具有相对很低的强度,归属于孤立的、具有较低格位对称性的Mn4+。Na2GeF6∶Mn4+样品的EPR谱与Na2SiF6∶Mn4+样品类似,其EPR信号也来自于具有较高和较低格位对称性的Mn4+离子。根据EPR信号积分强度,可推断在Na2GeF6∶Mn4+中处于较高格位对称性的Mn4+的数量减少、而处于较低格位对称性的Mn4+的数量增多。而在Na2TiF6∶Mn4+的EPR谱中可观察到许多信号,说明样品中含有不少于两种具有不同磁学性质的Mn4+。根据EPR信号积分强度可推断处于较高和较低格位对称性的Mn4+离子的数量几乎相当。根据三样品EPR积分强度所推断的处于较高和较低格位对称性的Mn4+离子相对数量变化趋势,与所观察的零声子线强度的变化趋势一致。因此,当样品中具有处于不同格位对称性的Mn4+离子时,可观察到多个EPR信号,其相对强度可用于表征Mn4+所处格位对称性。
多重超精细结构是由于Mn原子核的磁矩引起的,与Mn的价态无关[40]。特征六重超精细结构可用以证明Mn元素的存在;但文献报道EPR技术检测Mn价态时存在相互矛盾之处,比如文献[31,41]认为Mn3+和Mn5+离子对EPR静默,而文献[42]报道EPR谱中同时观察到了对应于Mn2+、Mn3+、Mn4+的信号且Mn4+和Mn3+产生与Mn2+类似的EPR谱。因此,使用EPR表征荧光粉中Mn的化合价时,最好结合其他手段验证。
3.5 阴极射线发光光谱
阴极射线发光(Cathodoluminescence,CL)指电子束激发发光材料引起的发光。电子束的电子能量通常在几千至几万eV,入射到发光材料中产生大量次级电子,离化和激发发光中心产生发光;与荧光光谱相似,CL也可表征多价态锰离子。Zhang等[28]在1 750 ℃下真空烧结6 h制备了Lu3Al5O12∶Mn2+透明荧光陶瓷,图7(a)、(b)所示为其在空气中1 400 ℃退火5 h后的CL光谱和CL图像。在电子束激发下所有晶粒均有发光现象(图7(b)),表明发光中心成功地分布在各晶粒中。CL光谱由3个波段组成,峰值位于588 nm处的强谱带源自Mn2+发光,峰值为666 nm处的谱带来源于Mn4+发光(639 nm发光应为Mn4+离子2E→4A2跃迁的反斯托克斯发射峰),700~900 nm范围内的宽带发射可能来自Mn3+。该CL结果表明,真空烧结所制备的Lu3Al5O12∶Mn2+陶瓷中,部分Mn2+离子经退火处理成功转变为Mn4+,但是同时也生成了Mn3+。图7(c)、(d)所示为在O2气氛下经1 500 ℃退火处理的Lu3Al5O12∶Mn荧光陶瓷的CL光谱和图像[29]。可以看出,其表现出单一的主峰波长为666 nm的CL发光,具有Mn4+离子2Eg→4A2g跃迁特征,与其PL谱一致,且Mn4+在颗粒中的发光很均一。表明在O2中退火比在空气中退火处理后,更有利于Mn2+充分转变为Mn4+。由图7可以看出,CL光谱可在微米至亚微米范围表征荧光材料中具有可见光发射的Mn4+和Mn2+离子,也能表征具有红外光发射的Mn3+离子。
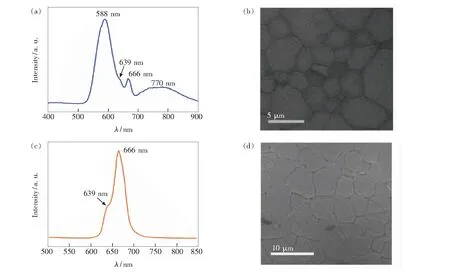
图7 Lu3Al5O12∶Mn透明陶瓷在空气气氛下经1 400 ℃退火处理后的CL光谱(a)和CL图像(b)[28]; Lu3Al5O12∶Mn透明陶瓷在O2气氛下1 500 ℃退火处理后的CL光谱(c)和CL图像(d)[29]。
3.6 X射线吸收近边结构谱
X射线与物质相互作用时,拥有足够能量的光子被吸收,将使得束缚电子发生跃迁。由于价带和导带底能级受到邻近配位原子的影响,吸收概率随能量的变化而显示出一种精细结构谱,其中包含原子的化学状态信息,例如价态、配位数、配位原子、键长等。X射线吸收精细结构谱(X-ray absorption fine structure,XAFS)分为两个区域:由低能光电子在配位原子做多次散射后再回到吸收原子与出射波发生干涉形成的X射线吸收近边结构(X-ray absorption near edge structure,XANES),其特点是强振荡;以及电离光电子被吸收原子周围的配位原子做单次散射回到吸收原子与出射波发生干涉形成的扩展X射线吸收精细结构(Extended X-ray absorption fine structure,EXAFS),其特点是振幅不大、似正弦波动。XANES对结合能更敏感,非常适合于价态确定;EXAFS反映分子环境的特性,例如配位数、配位离子以及它们之间的距离。XANES技术用于元素价态检测的优点有:适用于具有元素特异性的任何元素,浓度灵敏度低(低至10-6),并且适用于检测包括液体和粉末在内的任何形式的样品[43]。
通过比对XANES谱中样品MnK边与参比物MnK边的相对位置,可鉴定Mn离子在荧光粉中的化合价。Ye等[37]用XANES表征所合成的Mg2TiO4∶Mn4+红光荧光粉中Mn离子的价态。图8(a)所示为Mg2TiO4∶Mn4+及一系列参比化合物(MnO、Mn2O3、β-MnO2)的MnK边的XANES谱[37]。Mg2TiO4∶Mn4+样品的MnK边与β-MnO2的相似,因此表明Mn离子的氧化态为+4。Takeda等[44]以MnCO3为Mn源,以AlN、α-Al2O3、MgO为原料,高温固相法合成了Mn/Mg共掺γ-AlON绿光荧光粉。图8(b)所示为γ-AlON∶Mn,Mg和含Mn2+(MnO和MnCO3)、Mn3+(Mn2O3)、Mn4+(MnO2)参比化合物的标准化MnK边XANES谱[44]。γ-AlON∶Mn,Mg的XANES谱包含一个在6 537 eV处的前缘峰和一个在6 544 eV处的主峰。γ-AlON∶Mn,Mg的吸收边位于6 542.5 eV,介于MnO和MnCO3之间。因此,Mn在γ-AlON∶Mn,Mg中的价态推断为+2。
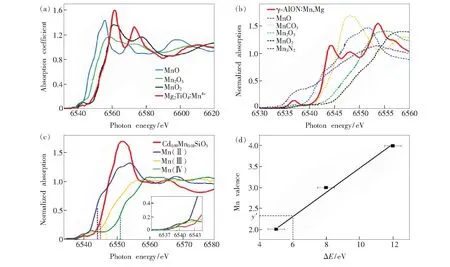
图8 (a)Mg2TiO4∶Mn4+的XANES谱(nMn/(nMn+nTi)=0.10%)及一系列参比物(MnO、Mn2O3、β-MnO2)的XANES谱[37];(b)γ-AlON∶Mn,Mg及MnO、MnCO3、Mn2O3、MnO2和Mn3N2等参比物的Mn K边XANES谱[44];(c)Cd0.99-Mn0.01SiO3和参比物MnO、Mn2O3和MnO2的Mn K边XANES谱,竖直虚线所指为各条谱线的吸收边;(d)Mn价态与Mn K边化学位移间函数关系(实线为线性拟合结果),图中也给出了Cd0.99Mn0.01SiO3样品中Mn离子的平均价态y′[45]。
此外,还可通过XANES图谱中样品吸收边与参比物吸收边的相对位置判断样品中不同价态离子的相对含量。Abreu等[45]以MnCl2·4H2O为Mn源,CdO和SiO2为原料,经高温固相法合成Cd0.99Mn0.01SiO3长余辉荧光粉,并用XANES对Mn价态进行表征。图8(c)所示为Cd0.99Mn0.01-SiO3和参比化合物MnO(Mn2+)、Mn2O3(Mn3+)和MnO2(Mn4+)的MnK边XANES谱[45]。如图8(c)插图所示,Cd0.99Mn0.01SiO3的XANES谱的前端与Mn2+相似,表明Mn主要以+2价存在于CdSiO3基质中。Cd0.99Mn0.01SiO3的吸收边位于6 545 eV,介于Mn2+(6 544 eV)和Mn3+(6 547 eV)之间,表明样品中还包含少量Mn3+。为进一步探究Mn2+与Mn3+的相对含量,以Mn价态与能量位移(ΔE)间的函数关系作图,示于图8(d)。其中,ΔE由公式(1)决定:
ΔE=Eo-EMn(K),
(1)
EMn(K)(6 539 eV)对应于金属态Mn原子的吸收K边,Eo为由参比物确定的实验值。用线性函数y=a+bΔE拟合数据,解得斜率b=(0.28±0.02),截距a=(0.6±0.2)。对Cd0.99Mn0.01SiO3来说,ΔE=6 eV(图8(c)),因此代入y=a+bΔE得到该样品中Mn的平均价态y′=(2.3±0.3)。这是由Mn2+和Mn3+同时存在所致,进而由公式(2)可以求出各自的比例:
y′=(2X2+3X3)/(X2+X3),
(2)
其中,X2和X3分别代表Mn2+和Mn3+的比例,X2+X3=1。由公式(2)得出,X2=0.72,X3=0.28,因此得到相对比例为Mn2+为72%,Mn3+为28%。
利用XANES谱表征荧光粉中锰离子化合价时,因其对Mn离子化学环境很敏感,参比物不同时,MnK边位置将有所不同。因此,应根据荧光粉中Mn离子的配位环境选择与其配位环境接近的化合物为参比物。
3.7 变温磁化率测试
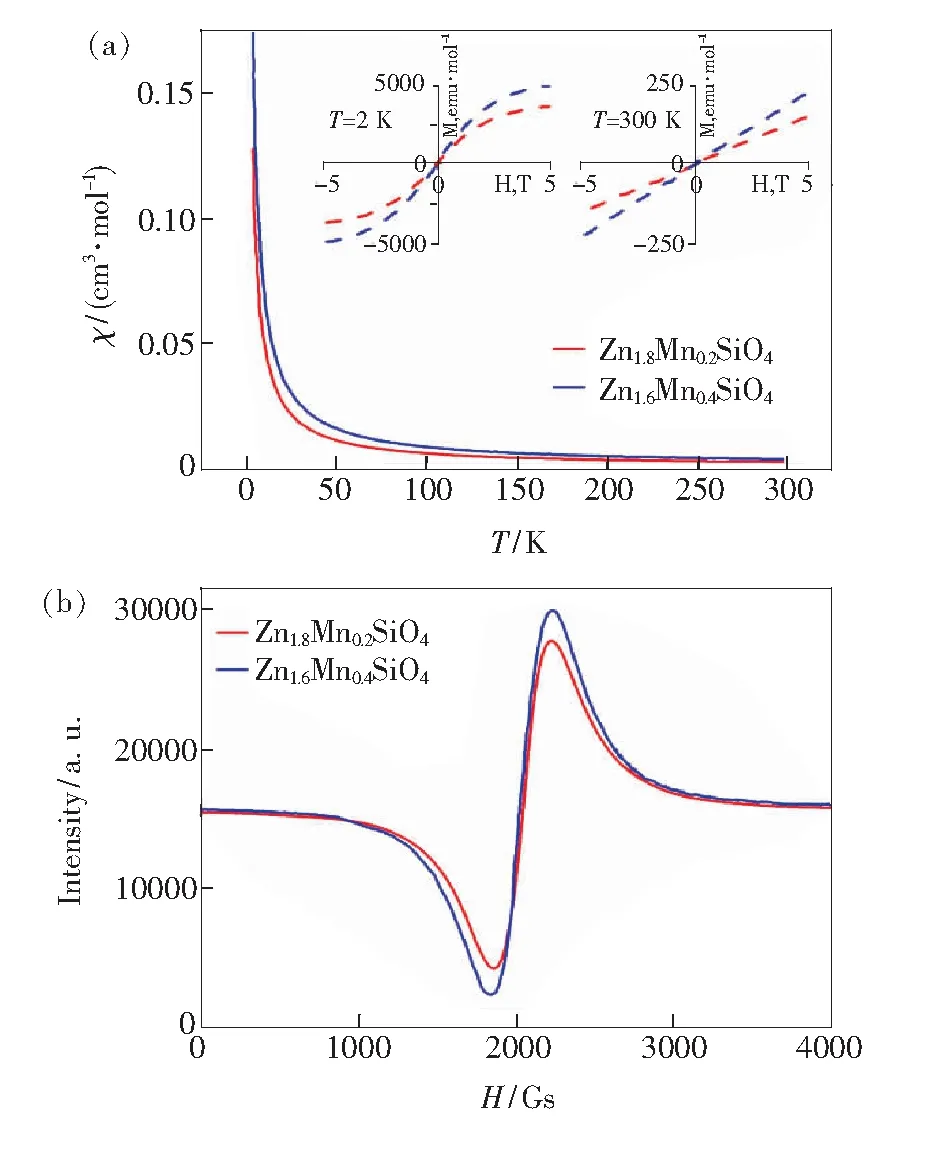
图9 (a)Zn1.8Mn0.2SiO4和Zn1.6Mn0.4SiO4样品的磁化率(χ)与温度的关系(插图所示为两样品在2 K和300 K时其磁化强度的场依赖性);(b)两样品的室温EPR图谱[46].
4 总结与展望
锰激活荧光粉中常存在多价态(常常是Mn4+/Mn3+/Mn2+)共存现象。多种方法可表征其所含锰离子价态,列于表1。其中,荧光粉颜色(体色及激发光下的荧光色)能大概反映所含锰离子价态信息。漫反射光谱测试便易性较强,同时适用于荧光猝灭及未猝灭样品,可定性表征多价态,但无法定量表征多价态锰离子间相对含量。荧光光谱可对能产生荧光信号的样品进行表征,可与漫反射光谱结合而定性表征Mn2+/4+离子。EPR谱仅可进行Mn2+/3+/4+定性表征,但荧光粉中锰离子EPR信号的影响因素多,需要在正确工作参数下测试才能够得到真实反映Mn离子价态或存在形式的EPR信号。此外,EPR信号不仅能反映锰离子价态信息,还能够一定程度地反映其配位环境、荧光粉中缺陷等的存在情况信息,这对于分析荧光粉发光性质很有益处。XPS谱表征样品表面的化学信息,且需要对所得XPS谱进行分峰处理;但由于样品中Mn含量低,其对应的Mn 2p信号弱,由分峰结果来定量表征各价态锰离子相对含量的可靠性需进一步研究。CL发光谱可对样品进行纳微米范围内的多价态研究,在微区范围的空间分布表征及材料内部(利用新鲜断面)表征方面有明显优势;其信号获得相对简单,可获得点/面的发光光谱与发光图像,可与扫描电子显微镜及能谱仪联用,进行微区形貌-组成-光谱的联合分析,在定性表征多价态锰离子时值得推广使用;但目前没有CL光谱定量表征多价态的文献报道,是否可以通过先建立一个工作曲线来实现定量表征多价态锰离子相对含量,需进一步研究。XANES谱需使用同步辐射光源,常难以获得测试机会;信号灵敏度高,可进行10-6级定量表征;但需选择与样品化学环境相近的多种参比样品进行测试,多价态锰离子的相对含量结果与所选参比物密切相关。变温磁化率谱需进行从极低温开始的变温实验,实验条件相对苛刻;可区别Mn2+/Mn3+,但难以准确定量表征多种价态之间的相对含量。漫反射光谱、荧光光谱、XPS谱、CL发光光谱和XANES谱不仅是锰离子而且是其他发光离子价态分析的可用手段,而EPR谱和变温磁化率谱利用锰离子未充满d轨道电子所表现出的磁学性质,是锰离子价态表征相对独特的技术手段。在定性表征方面,漫反射光谱、荧光光谱是表征多价态锰离子的有效手段,可与CL发光光谱联用表征Mn4+/Mn3+/Mn2+离子;荧光光谱可与EPR谱联用,表征荧光粉中Mn4+离子配位八面体的畸变程度。在定量表征方面,XANES谱可定量表征Mn4+/Mn3+/Mn2+离子;若不使用参比物,则可通过变温磁化率谱与EPR谱联用方法进行定量表征。

表1 锰离子激活荧光粉中锰离子价态表征手段
最后,定性或定量Mn激活荧光粉中锰离子价态的最终目的之一是为了探明影响其价态的因素以及控制掺杂价态为预期价态。因此,本文进一步对影响锰离子掺杂价态的因素及其价态控制方法归纳如下:
(1)烧结气氛:高温固相反应时,一般氧化气氛热处理有利于Mn4+形成而还原气氛热处理有利于Mn2+形成。
(2)晶体结构:Mn4+在八面体配位环境中稳定,因此在研发Mn4+激活红光荧光粉时,应寻找含有大量八面体配位的化合物作为基质。Mn2+可存在于四面体或八面体配位,分别受弱和强晶体场作用而发绿光或橙红光。Mn3+可在四面体格位稳定存在。
(3)所取代的离子的价态和有效离子半径:通常情况下,与被取代离子的价态越接近,造成的电荷不平衡越小,越易得到所需价态;与被取代离子的有效离子半径越接近,造成的晶格畸变越小,越易保持价态稳定。
(4)电荷补偿剂:加入电荷补偿剂如Mg2+,可有效促进Mn4+在铝酸盐基质中的发光,因为电荷补偿机制Mg2++Mn4+→2Al3+促进了更多锰离子以4+形式存在。
(5)费米能级在禁带中的位置:费米能级越靠近价带,即(εf-EVBM)越小,越易形成Mn4+;反之越易形成Mn2+。Chen等[33]通过第一性原理计算从电子结构角度解释了共掺杂剂Li+、Mg2+、Na+和Si4+对Mn激活铝酸盐(α-Al2O3和BaMgAl10O17)发光性能的影响。计算结果表明,当费米能级(εf-EVBM)低于1.86 eV时,Mn4+形成能最低;费米能级高于1.86 eV时,Mn4+变得不稳定并可转变为Mn3+;费米能级高于3.78 eV时,Mn2+最稳定。Na+、Mg2+共掺杂可形成空穴型缺陷,降低费米能级的位置而提高Mn4+的稳定性;Li+共掺杂不仅形成空穴型缺陷LiAl,而且形成间隙缺陷Lii,后者可提供电子给导带,而降低Mn4+的稳定性。Si4+共掺杂可形成SiAl和SiAl+VAl复合缺陷,前者降低Mn4+的稳定性,后者作为空穴缺陷提高Mn4+的稳定性。
笔者认为,为了研发高效白光LED用Mn4+激活红光荧光粉,可采取多种方法来促进所掺杂锰离子以Mn4+形式存在而抑制其他价态的影响:选取具有较多八面体配位结构的化合物作为基质;基质化合物结构中应尽量不含或少含可容纳Mn3+或Mn2+的四面体配位结构等;取代离子应电荷平衡、半径匹配度高;控制Mn的掺杂浓度在适量水平;进行电荷补偿剂离子的共掺;在氧化气氛下煅烧合成。
猜你喜欢
学与玩(2022年6期)2022-10-28
上海金属(2022年5期)2022-09-26
陶瓷学报(2021年5期)2021-11-22
陶瓷学报(2020年6期)2021-01-26
贵州农机化(2020年3期)2020-12-24
辽河(2020年12期)2020-01-05
活力(2019年17期)2019-11-26
中国计量大学学报(2018年2期)2018-07-12
中国测试(2018年4期)2018-05-14
植物营养与肥料学报(2011年2期)2011-10-26
