
Association between human leukocyte antigen gene polymorphisms and multiple EPIYA-C repeats in gastrointestinal disorders
2020-09-23 10:03SuatSaribasSuleymanDemiryasErkanYilmazOmerUysalNurayKepilMehmetDemirciReyhanCaliskanHarikaOykuDincSeherAkkusNesrinGareayaghiSahraKirmusaogluDogukanOzbeyHrisiTokmanSerdarKoksalIhsanTasciBekirKocazeybek
World Journal of Gastroenterology 2020年32期
Suat Saribas, Suleyman Demiryas, Erkan Yilmaz, Omer Uysal, Nuray Kepil, Mehmet Demirci, Reyhan Caliskan, Harika Oyku Dinc, Seher Akkus, Nesrin Gareayaghi, Sahra Kirmusaoglu, Dogukan Ozbey, Hrisi B Tokman, Serdar S Koksal, Ihsan Tasci, Bekir Kocazeybek
Abstract
Key words: Human leukocyte antigen; Helicobacter pylori; Gastric cancer; Duodenal ulcer; EPIYA; CagA
INTRODUCTION
Gastric cancer (GC) is the third most common cause of death among all cancer types (8.2% of all cancers, 2018, World Health Organization)[1]. GC has been closely associated withHelicobacter pylori(H. pylori) infections, which generally cause mild gastrointestinal symptoms. However, in a few infected patients, they may progress to peptic ulcer (PU) (10%-15%) or GC (1%-3%)[2].
Generally,H. pyloriinfections cause gastric inflammation and a chronic inflammatory response that result in progressive mucosal damage. Eventually, the gastric mucosa transforms into metaplastic and dysplastic epithelia, which leads to gastric adenocarcinomas[3]. The development of GC is suggested to be related to the interactions between bacterial virulence factors, host genetic factors such as the human leukocyte antigen (HLA) gene, the immune response of the host, and environmental factors such as diet and smoking[4]. Some of the host factors may be related to polymorphisms genes such as HLA genes, which regulate the strength of the inflammatory response and influence the probability of specific clinical results[5].
CagA is among the most important virulence factors specific toH. pyloriand is delivered into the gastric epithelium cells by the type IV secretion system (T4SS) of the bacterium. It is involved with intracellular signal transduction pathways and leads to the malignant transformation of gastric epithelial cells[6]. CagA subtypes are defined by the EPIYA variants (Glu-Pro-Ile-Tyr-Ala) in their C-terminal region, and there are four types of EPIYA motifs: EPIYA-A, -B, -C, and -D[7].H. pyloriisolates from East Asia are associated with a higher incidence of GC and contain EPIYA A-B-D motifs. However, in Western countries, GC cases have EPIYA A-B-C motifs.H. pyloristrains with multiple EPIYA-C repeats have higher phosphorylation capacity and SHP-2 binding affinity and are significantly associated with GC[8].
HLA class I and II proteins bind to bacterial antigen peptides or tumor proteins and present these peptides and proteins to T cells, which leads to the differentiation of T cells into cytotoxic or helper T cells. In HLA gene polymorphism, a variety of alleles may occupy the same locus. A limited number of studies have focused on the relation between gastrointestinal pathologies such as the development of GC risk and HLA polymorphisms in different geographic regions and populations[9,10,11].
In addition to HLA gene polymorphisms, the virulence factors ofH. pyloristrains may also be related to the intensity of the inflammatory response[3]. The limited studies on HLA polymorphisms have focused on MHC class II and specifically the relation of HLA polymorphisms and the development of GC risk[10,12,13]. In these studies, the following HLA class II polymorphisms were suggested to be associated with the development of GC risk: DQB1*03, HLA-DRB1*04, and HLA-DQA1*01/*03[14,15].
Reported HLA allele frequencies related to GC pathologies withH. pyloripositivity have been contradictory among different ethnic groups. The regional and ethnic differences are very important for the association of HLA gene polymorphism and GC risk. These contradictory results may be attributed to factors such as differences in populations, research designs, environmental factors,H. pylorivirulence factors, and host genetic factors, such as polymorphisms of HLA alleles. For example, the HLADQB1*0301 allele was positively associated with GC in Caucasian populations but negatively associated with it in Taiwanese populations[13,16]. This HLA polymorphism had no effect in Japanese populations[17]. HLA-DQB1*0401 82 and *0602 alleles increase the GC risk in European and Indonesian populations[9,18].
To the best of our knowledge, no studies have compared GC and duodenal ulcer (DU) cases with controls in regard to HLA allele frequencies in terms of differentiation by the CagA+ multiple (≥ 2) EPIYA-C repeat numbers. Therefore, we aimed to investigate the allele frequencies of HLA class I and II in a patient group [H. pylori(+) GC and DU patients] and compared the results to those of a control group [H. pylori(+) non-ulcer dyspepsia (NUD) and asymptomaticH. pylori] in terms of CagA+ multiple EPIYA-C repeats for the first time in a Turkish population.
MATERIALS AND METHODS
Study design and patients
This case–control study was conducted between July 10, 2014 and November 9, 2017. The patient group comprised 94 patients, including 44 (46.8%) GC and 50 (53.2%) DU patients with 58 (61.7%) males, 36 (38.3%) females, a mean age of 49.6 years, and age range of 19–79 years. The control group comprised 86 individuals including 50 (58.1%) NUD patients and 36 (41.9%) people with asymptomaticH. pylori. This group had 30 (34.9%) males, 56 (65.1%) females, a mean age of 47.3 years, and age range of 18–86 years. All of the GC + DU patients and the NUD + asymptomaticH. pylorimembers of the control group members hadH. pylori.
The NUD + AsymptomaticH. pyloricontrol group was matched with the GC + DU patient group according to the age and gender distribution of the patient group (P> 0.05). Blood samples for the genotyping of HLA alleles were collected when obtaining biopsy samples (from the corpus and antrum) on the same day. The antrum and corpus biopsy specimens were stored and used in molecular studies.
We excluded patient and control group individuals with autoimmune diseases and who were under 18 years old, had previous gastric surgery andH. pylorieradication treatment with antibiotics, antisecretory drugs and bismuth salts, in the month prior to sampling. The study was reviewed and approved by the Clinical Research Ethics Board of Istanbul University-Cerrahpasa, Cerrahpasa Faculty of Medicine (No. 83045809/32-38/A-15/2014). The study was also conducted according to the standards of the Declaration of Helsinki. All study participants or their legal guardians provided informed written consent prior to the study.
Polymerase chain reaction analyses
H. pyloriDNA extractions were performed using the antrum and corpus biopsy specimens. Genomic DNA extraction (Real Genomics Quality Nucleic Acid/ Purification system; RBC Bioscience Laboratories, Taipei, Taiwan) and QIAamp DNA mini prep kits (Qiagen, Hilden Germany) were used.
UreC gene detection in H. pylori
AnH. pylori-QLS 1.0 kit (Fluorion, Iontek, Istanbul, Turkey) was used for the detection of 156 bp of theureCgene inH. pyloriDNA extractions[19].
Amplification of the H. pylori cagA gene
Primers reported in related studies were used for the detection of theH. pylori cagAgene (349 bp) (Table 1)[20,21]. The polymerase chain reaction (PCR) cycles were as follows: Denaturation at 95 ºC for 2 min, followed by 45 cycles of 95 ºC for 30 s, 45 s at 53 ºC, and 45 s at 72 ºC. The final elongation was done for 5 min at 72 ºC.
Molecular studies for the typing of EPIYA motifs
The amplification ofH. pyloriDNA for EPIYA motifs was done using the forward primer cagA28F and reverse primers cagA-P1C, cagA-P2CG, cagA-P2TA, and cagAP3E (Table 1)[22]. In the PCR assay, the protocol steps were as follows: Initial denaturation step, one cycle at 95 ºC for 2 min, 50 cycles at 95 ºC for 30 s, 57 ºC for 45 s, and 72 ºC for 35 s; and a final extension at 72 ºC for 5 min. After the PCR amplification, PCR products were sequenced bidirectionally using a Sequence Reagent Mix kit with an ABI Prism (310) analyzer (Applied Biosystems, United States).
Empty-site PCR
An empty-site-positive PCR assay was used to confirm the EPIYA-negativeH. pyloristrains[23]. To confirm cagPAI in all of the strains, amplification was performed with two primers (forward 468 HP519 and reverse 496 HP549 primers of the reference HP519 and HP549H. pyloristrains; Table 1, cag empty PCR). In the PCR assay, the protocol steps were as follows: Initial denaturation at 95 ºC for 2 min, 40 cycles at 95 ºC for 30 s, 57 ºC for 30 s, and 72 ºC for 20 s; followed by a final extension at 72 ºC for 5 min.
Blood collection, DNA extraction, and HLA sequence-specific oligonucleotide typing
Whole blood samples (10 mL) were collected from the patient and control group cases during a biopsy procedure. An EZ1 DNA extraction kit (Qiagen, Germany) was used in a DNA isolation device (Bio Robot EZ1; Qiagen, Germany) for the DNA isolation procedure from 3-mL blood samples collected in tubes containing ethylenediaminetetraacetic acid. Isolated DNA samples were stored at -70 °C until laboratory studies. HLA typing at low levels (2 digits) of HLA-A, HLA-B, HLA-C, and HLA-DQ alleles were done with a Luminex 100/200 instrument with sequencespecific oligonucleotide (SSO) probes bound to color-coded microbeads. LIFECODES SSO HLA typing kits (Lifecodes, Immucor, Germany) were used for the typing of HLA-A HLA-B, HLA-DRB1, and HLA-DQA1/B1. This typing test is a reverse sequence-specific oligonucleotide (rSSO) DNA typing assay using SSO probes and color-coded microspheres.
The PCR mixture was composed of 15 μL of lifecodes Master Mix, 200 ng ofgenomic DNA, and 2.5 U Taq polymerase in a final volume of 50 μL. In the PCR assay, the steps were as follows: Initial denaturation step at 95 °C for 5 min; 40 cycles including 8 cycles at 95 °C for 30 s, 60 °C for 45 s, and 72 °C for 45 s, followed by 32 cycles at 95 °C for 30 s, 63 °C for 45 s, and 72 °C for 45 s, and a final extension step at 72 °C for 15 min. The hybridization steps were as follows: Initial step at 97 °C for 5 min followed by 30 min at 47 °C for 30 min and at 56 °C for 10 min with 15 μL of probe mix and 5 μL of PCR product. The obtained samples were diluted with 170 μL of 1:200 prediluted streptavidin-phycoerythrin solution and analyzed by a Luminex 200 system (Luminex Corp. United States). The obtained HLA patterns were compared with ah HLA sequence database (Database for IMGT/HLA Sequence, 3.11.0) using the MatchIT DNA program.

Table 1 PCR primers
Statistical analyses
Hardy-Weinberg (H-W) equilibrium and Linkage Disequilibrium (LD) were examined for HLA-A, HLA-B, HLA-C, HLA-DR1, HLA-DQA1 and HLA-DQB1 allele polymorphisms[24]. Genepop software version 4.7 was used to calculate the Hardy–Weinberg equilibrium and LD. H–W equilibrium was present forP-values > 0.05. The allele frequencies of the GC + DU patient and the NUD + AsymptomaticH. pyloricontrol groups and subgroups were compared by the chi-squared (χ2) test and Fisher’s exact test (Tables 2-5). CorrectedPvalues (Pc) were calculated by multiplying with the allele numbers (A = 15, B = 26, DRB1 = 12, DQA1 = 6, and DQB1 = 5) in each locus by Bonferroni correction. Multivariate logistic regression (enter method) was used to assess the relation of HLA alleles and the risk of GC and DU development in terms of CagA+ multiple EPIYA-C repeat numbers (Table 6).P< 0.05 was used to determine significance. SPSS 25.0 (IBM Corporation, Armonk, NY, United States) was used for the analyses.
RESULTS
Sixty-four HLA alleles (41 for class I and 23 for class II) were found between the 94 HLA alleles tested in all of the groups (Table 7). The maximum numbers of alleles in the GC + DU patient group were 52 for HLA-A*02 (27.6%), 40 for HLA-B*35 (21.3%), 36 for HLA-DRB1*13 (19%), 76 for HLA-DQA1*01 (40%), and 52 for HLA-DQB1*06 (27.6%) (Figure 1). The maximum numbers of alleles in the NUD + AsymptomaticH. pyloricontrol group were 20 for HLA-A*03 (11.6%), 10 for HLA-B*50 (5.81%), 24 for HLA-DRB1*04 (13.95%), 54 for HLA-DQA1*05 (31.4%), and 64 for HLA-DQB1*03 (37.2%) (Figure 2). Among GC cases, the genotype frequencies of HLA- B, DQA1 and DQB1 loci were in H-W equilibrium. Among DU cases, all of the genotype frequencieswere in H-W equilibrium. Moreover, Among NUD cases, all of the genotype frequencies in H-W equilibrium except HLA-DQB1 cases and in asymptomaticH. pyloricontrol group, all of the genotype frequencies were in H-W equilibrium except HLA-A cases. H-W equilibrium is only valid with a very large sample size and therefore, we suggest that some of our genotype frequencies are not in H-W equilibrium. The deviations from H-W equilibrium is larger at small sample sizes like this study and smaller at large sample sizes. HLA-DR1*13-HLA-DQA1*01-HLADQB1*06 haplotype frequency (10/44, 22% for GC; 2/50, 4% for DU, 2/50, 4% for NUD and 0 for NGIS) showed LD and had an odds ratio (OR) value as 6.143 (95%CI: 1.33-28.31,P= 0.0027) in the comparison of patient and control group cases. Moreover, HLA-DR1*13-HLA-DQA1*01-HLA-DQB1*06 haplotype frequency compared between GC and DU cases and OR was detected as 10.58 (95%CI: 2.27-49.34).
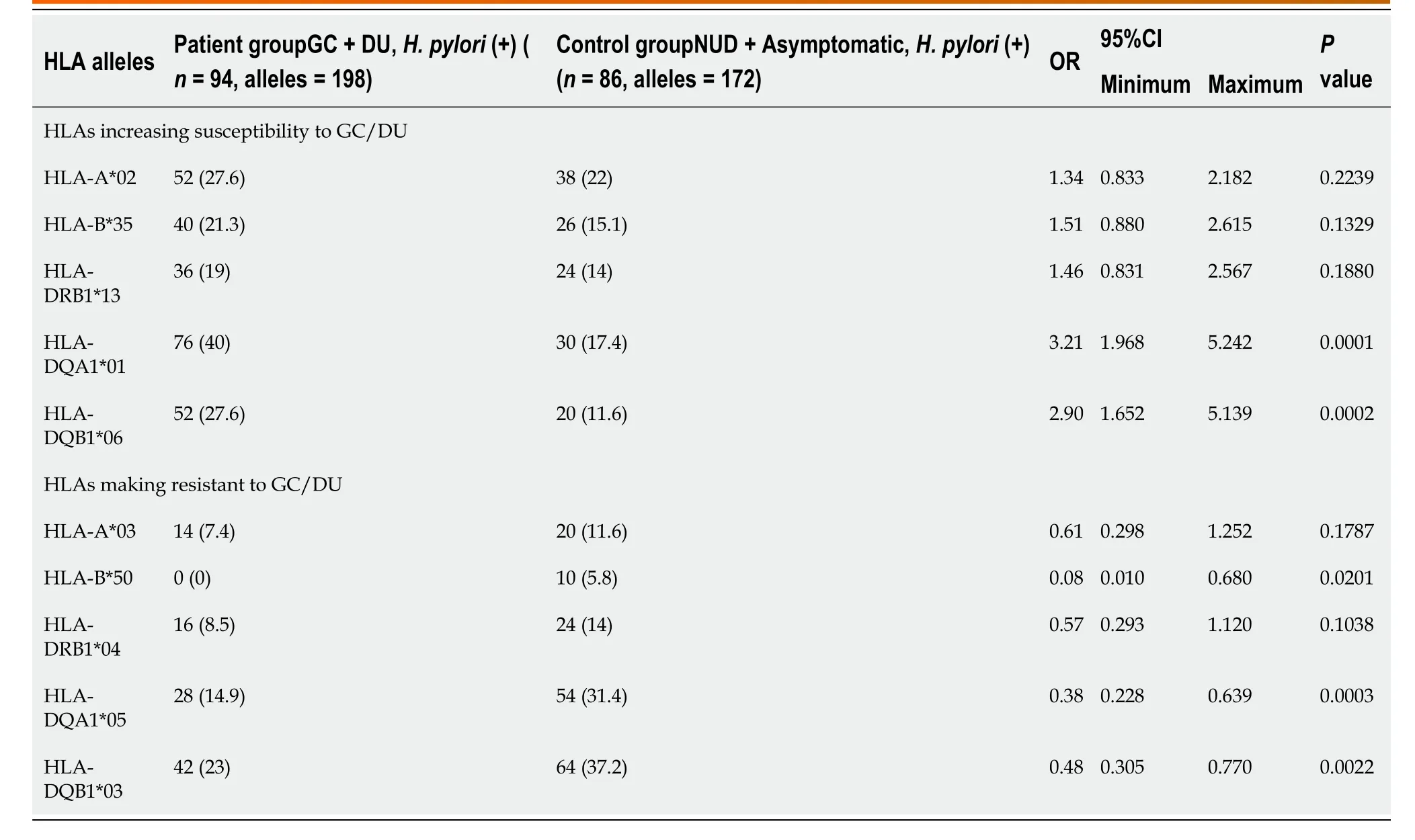
Table 2 Comparison of human leukocyte antigen alleles predisposing susceptibility and resistance to gastric cancer/duodenal ulcer in patient and control groups, n (%)

Table 3 The comparison of human leukocyte antigen alleles which increase or decrease the gastric cancer risk in gastric cancer subgroup cases in terms of CagA+ (≥ 2) EPIYA-C, n (%)
When comparing the prominent alleles detected, only HLA-DQA1*01 (OR: 3.211,P= 0.0001) and HLA-DQB1*06 (OR: 2.906,P= 0.0002) were significantly higher in theGC + DU patient group. The values also stayed significant after Bonferroni correction (DQB1*06:Pc= 0.001; DQA1*01:Pc= 0.0006). The prominent alleles in the NUD + AsymptomaticH. pyloricontrol group were HLA-B*50 (OR: 0.086,P= 0.02), HLADQA1*05 (OR: 0.384,P= 0.0003), and HLA-DQB1*03 (OR: 0.485,P= 0.0022), but after Bonferroni correction for multiple comparisons, the changes were statistically significant for only HLA-DQA1*05 (Pc= 0.0018) and HLA-DQB1*03 (Pc= 0.011) and not HLA-B*50, (Pc= 0.52) (Table 2).

Table 4 The comparison of human leukocyte antigen alleles which increase or decrease the duodenal ulcer risk in duodenal ulcer subgroup in terms of CagA+(≥ 2) EPIYA-C, n (%)

Table 5 The comparison of human leukocyte antigen alleles which increase or decrease the gastric cancer/duodenal ulcer risk in gastric cancer and duodenal ulcer subgroups in terms of CagA+(≥ 2) EPIYA-C, n (%)

Table 6 Results of logistic regressions analysis according to the variables in patient group (gastric cancer and duodenal ulcer cases)
Multiple EPIYA-C repeat numbers and CagA positivity were found in 40 (42.5%) subjects in the GC + DU patient group. Multiple EPIYA-C repeats were observed in 26 (59%) GC subgroup cases and 14 (28%) DU subgroup cases in the GC + DU patient group, 2 (2.3%) NUD cases, and no asymptomaticH. pyloricases. Other EPIYA motifs and their numbers are shown in Table 8. It was not possible to perform statisticalanalysis due to small number of positive cases with CagA+ multiple EPIYA-C repeats in the NUD+ AsymptomaticH. pyloricontrol group (n= 2). Instead, we compared two groups without using any criteria with regard to HLA alleles.

Table 7 Frequency of detected human leukocyte antigen class I alleles and the class II alleles in patients with cancer and ulcer and in the control groups, n (%)

HLA: Human leukocyte antigen.
Thus, we used the HLA-DQB1*06, HLA-DRB1*13, HLA-B*35, HLA-DQA1 and HLA-A*02 alleles (i.e., the alleles with the maximum numbers in the GC + DU patient group) to investigate their effects on GC/DU in terms of the CagA+ multiple EPIYA-C repeats, which is suitable for our purposes. First, we compared alleles in GC cases in terms of CagA+ multiple EPIYA-C repeat numbers. Only the HLA-DQB1*06 allele (OR: 0.37, 95%CI: 1.149-0.942,P= 0.0369) was significantly associated with GC, but after Bonferroni correction, there was no significant association between DQB1*06 and GC (DQB1*06,Pc= 0.1845).
The HLA-DQA1*01 allele had a high ratio in the multiple EPIYA-C repeat group, but the univariate analysis did not show any significant association. None of the selected alleles were significantly higher in the GC cases in terms of CagA+ multiple EPIYA-C repeats (Table 3). Using the same criterion, we also compared allele frequencies in the DU cases, but none of the HLA alleles were significantly higher (Table 4). When we compared selected allele frequencies in the GC and DU cases together using this criterion, again, none of the HLA alleles were significantly higher (Table 5). HLA-DR1*13-HLA-DQA1*01-HLA-DQB1*06 haplotype frequency was 4/24, 16% and 2/12, 16% for GC and DU cases with multiple EPIYA-C repeat, respectively. No difference was detected between GC and DU cases in terms of multiple EPIYA-C repeat.
Multivariate logistic regression analyses were carried out for the risk of GC and DU development alone and both subcases of the GC + DU patient group combined involving HLA-DQB1*06, HLA-DRB1*13, HLA-A*02, HLA-DQA1*01, and HLA-B*35 alleles, and none of the alleles were detected as independent risk factors for the risk of GC and DU development in terms of CagA+ multiple EPIYA-C repeats. However, a multivariate logistic regression analysis was done without any specific criteria using only the significantly different detected alleles between GC + DU patient and the NUD+AsymptomaticH. pyloricontrol groups. The results showed that HLA-DQA1*01 (P= 0.004, OR = 1.848, 95%CI, 1.215-2.811), HLA-DQB1*06 (P= 0.009, OR = 1.821, 95%CI, 1.163-2.850), and HLA-A*02 (P= 0.04, OR = 1.579, 95%CI, 1.021-2.442) were risk factors for the development of GC and DU (Table 6).

Table 8 The distribution of EPIYA motifs for study and control groups, n (%)

Figure 1 The representations of the highest human leukocyte antigen allele frequencies (%) in the gastric cancer + duodenal ulcer patient group when compared to non-ulcer dyspepsis patients + asymptomatic Helicobacter pylori control group. HLA: Human leukocyte antigen; H. pylori: Helicobacter pylori.
DISCUSSION
The development of GC and PU or DU is influenced by the virulence factors ofH. pylorialong with the host’s genetic, epigenetic, and environmental factors. A variety of clinical consequences ofH. pyloriinfection may arise depending on the variability of host response to the specific virulence factors ofH. pylori[7]. For example, genes coding HLA class II molecules (HLA-DP/DQ/DR) may have genetically variable coding loci that lead to HLA gene polymorphisms. Therefore, specific HLA class II alleles were hypothesized to be related to the risks of some gastroduodenal malignancies such as GC and DU or PU development in patients withH. pyloriinfection.
In the literature, there are only traditional comparison studies using onlyH. pyloripositivity for the association between HLA gene polymorphisms and the diseases caused byH. pyloriinfections. However, we focused on CagA+ multiple EPIYA-C repeats for the comparison of our study groups for HLA alleles for the first time in Turkey. The incidence of GC in Turkey is higher than in Eastern countries and lower than in Western countries at 5.7 and 9.6 cases per 100000 people for women and men, respectively. The mean age of occurrence is 56 years. It is the second and third leading cause of cancer-related deaths in men and women in Turkey, respectively[25].

Figure 2 The representations of the highest human leukocyte antigen allele frequencies (%) in the (%) in the non-ulcer dyspepsis patients + Asymptomatic Helicobacter pylori control group when compared gastric cancer + duodenal ulcer patient group. HLA: Human leukocyte antigen; H. pylori: Helicobacter pylori.
The higher incidence in Turkey is mainly associated with dietary factors, and the differences in incidence are especially significant in the central, northeastern, and eastern regions of the country. Salt is commonly used for food preservation, and wood charcoal with dried cow dung is commonly used for cooking in these regions, which are known to have carcinogenic effects on food. Another important factor for the development of GC isH. pyloriinfections[25].
Studies have shown an association between HLA gene polymorphisms and autoimmune diseases, and in genetically susceptible individuals, persistent bacterial infections can lead to autoimmune responses (e.g., HLA-DR4-restricted autoimmune chronic synovitis following Lyme disease)[26]. While evaluating the effects ofH. pyloriinfection on the pathogenesis of GC, the relationship between bacteria and the host should be considered becauseH. pyloriinfections may cause strong immune responses by causing the secretion of cytokines from the epithelial cells and gastric mucosa infiltration with neutrophils, macrophages, and lymphocytes. After the interaction ofH. pyloriwith dendritic cells in luminal and subepithelial regions, dendritic cells may transform naive T cells into immunosuppressive Treg cells, and consequently, developed Th1 and Th17 cells may cause atrophic gastritis, epithelial hyperplasia, and intestinal metaplasia[27].H. pyloriinfections tend to cause chronic inflammation, which can increase an individual’s risk for the development of GC.
A commonly seen (90%) type of GC is adenocarcinoma, which is known to originate from the epithelial cells in chronic inflammation states[28]. Moreover, cytokines, which are the effector cells of inflammatory responses, may regulate a variety of immunologic events, including the inflammation, proliferation, and differentiation of epithelial cells. In the progression of gastric carcinogenesis, initially,H. pyloristrongly induces specific cytokines, but the immune response generally is not sufficient to clear theH. pyloriinfection completely from the human epithelial cells. As a result, chronic inflammation may occur[29].
Consequently, the tissue damage increases along with parietal cell atrophy and may progress to dysplasia and GC through the combined effects of the various factors of the host and the environment. A subtype of T cells, Th17 cells, and their associated inflammatory cytokines, interleukin (IL)-17A, IL-23, and IL-1β, have important roles in the development of GC, colorectal cancer, ovarian cancer, and hepatocellular carcinoma. Cytokine IL-23 plays a major role in the primary activation of IL-17A. Th17 responses are reported to be increased duringH pyloriinfections. The IL-17/IL-23 axis is believed to have an important role in the progression of chronic inflammation and related pathologies like gastric neoplasms[30].
Other than their main roles in chronic inflammation in gastric epithelial cells, cytokines also have specific polymorphisms in their genes. Polymorphisms in cytokine genes may modify the effect of gene-environment interaction and increase the degree of cytokine expression in the promoter regions of the genes. Polymorphisms in genes coding various cytokines such as IL-1β, IL-1Ra, IL-8, IL-10, and tumor necrosis factor-α are also suggested to be associated with the risk of GC. The IL1RN2 allele polymorphism is related to the risk of GC[31]. Wuet al[32]found a relation between IL-17F, A7488G and GC, and Felipeet al[33]also reported a relation between IL-8 (rs4073)–251A/T gene polymorphism and GC development.
Some virulent factors ofH. pyloriseem to be associated with GC risk, including vacuolating cytotoxin A, cytotoxin associated antigen A, DU promoting gene protein A, and outer inflammatory protein with blood group antigen binding adhesins. Moreover, thecagAgene ofH. pyloristrains with multiple EPIYA-C repeats and EPIYA-D motif in itscagAgene are suggested to increase the risk of GC development. However, the role of host polymorphisms and the virulence factors ofH. pyloriin the risk of GC development varies among regions and ethnicities[31].
Several studies suggest that atrophic gastritis and GC risk are increased by CagApositiveH. pyloristrains. An association has been reported between multiple EPIYA-C phosphorylation sites and GC. In a recent meta-analysis including 23 studies, Liet al[8]evaluated the association of EPIYA motifs and gastroduodenal pathologies. They concluded that the EPIYA-D motif was significantly related to GC risk, and multiple EPIYA-C motifs were related to PU and DU in Asia countries.
Conversely, in the United States and Europe, multiple EPIYA-C motifs were commonly associated with GC risk. Multiple EPIYA-C repeats cause stronger binding of CagA to SHP-2 than a single EPIYA-C. Multiple EPIYA C repeats are associated with a higher risk of GC[34,35]. The functionality of CagA is increased with multiple EPIYA-C phosphorylation sites and is involved in cellular phenotypic changes. Therefore,H. pyloristrains with multiple EPIYA-C sites are related to the risk of GC.
We could not find any studies specifically evaluating the interaction between EPIYA-C repeats, HLA alleles, and gastric pathologies. In our study, there were only two cases in the control group with CagA+ multiple EPIYA-C repeats, and it was not possible to make a comparison as the number was too low and our criteria were not met. HLA-DQA1, HLA-DRB1*13, HLA-A*02, HLA-B*35, and HLA-DQB1*06 alleles were shown to contribute to the susceptibility to GC and DU and were used for a comparison between GC and DU subgroups of patients. However, we did not compare the higher HLA alleles detected in the control group as it was not possible to determine an HLA allele with a protective effect without including a control group.
In the comparison within the GC subgroup, due to our criterion, only the HLADQB1*06 allele was significantly low in the GC subgroup without EPIYA C repeats, but the difference was not statistically significant after Bonferroni correction. The higher frequency of the HLA-DQB1*06 allele suggests that this allele remains influential even in GC cases without multiple EPIYA-C repeats. With the presence of the HLA-DQB1*06 allele, we can suggest that mechanisms other than multiple EPIYAC repeats may contribute to the development of GC. On the other hand, in the GC subgroup, the number of HLA-DQA1*01 alleles was high in the multiple EPIYA-C repeats group, but the result was not significant after the univariate analysis.
In the DU subgroup, none of the alleles were found to be significantly predominant in terms of frequencies. In addition, when the analyses were repeated while including GC and DU subgroups together, none of the HLA alleles were shown to either effective or protective. A multivariate logistic regression analysis was performed for GC and DU subgroups alone and together, and none of the alleles were detected as independent risk factors. We then performed a multivariate logistic regression without including any discriminative criterion and only using the significantly different detected alleles between patient and control groups. As a result, HLA-DQA1*01, HLADQB1*0601, and HLA-A*2 alleles were found to be independent risk factors for the risk of GC and DU.
There are no previous studies to compare our results based on the discrimination criteria for the selection of HLA alleles. Our results partly coincided with those of a meta-analysis of Asian populations by Wanget al[36]. In that study, the susceptibility genes forH. pyloriinfection were reported as DQB1*0401, DQA1*0103, and DQA1*0301. Garza-Gonzálezet al[37]reported that the HLA-DQA1*0503 allele served as an independent protective factor for GC. In our study, we also detected HLADQA1*05 as a protective allele.
目前,我们在学校教育方面,也存在许多不完善的地方。一些学校为追求升学率,忽视了对青少年的青春期教育。一些学校对成绩好的同学呵护有加,而对成绩差的学生则放任自流。有的学校甚至对差生采取“停课”或“开除”等措施。不少学生流入社会后,成了“问题少年”。
Similar to our results, Quinteroet al[9]reported that the DQB1*0602 allele increased the risk of GC risk in a Southern European population infected withH. pylori. On the other hand, the results of several case–control studies conducted with a Japanese population were contradictory. In contrast to our results, Azumaet al[38]found that the number of DQA1*0102 alleles was significantly lower in their study group ofH. pyloriinfected patients with GC than in control subjects. However, our data did not support their findings for the DQA1* alleles.
In a study by Herrera-Goepfertet al[39], patients with GC displayed a high frequency of HLA-DQA1*0601, similar to our results. After our univariate analysis, DQA1*01 and HLA-DQB1*06 alleles were found to be positively associated with GC and DU, and DQB1*03 was found to be negatively associated with DU. These results are consistent with those of Herrera-Goepfertet al[39], Wuet al[13], and Quinteroet al[9].
3.假设单件化妆品的不含税价为P,买家购买套装相对于分别购买套装内所含产品的可享优惠率为R,则套装化妆品的不含税价为2P(1-R)。令,P≤2000,2P(1-R)>2000。 根据自网易考拉中选取的100组不参与活动打折的单件和套装化妆品样本,以“优惠率=[单件价-(套装价/套装所含件数)]/单件价”计算得知,优惠率R大多集中在0.15%-1.75%。
The reason for the conflicting results may be the different methods used in HLA typing, ethnicities, and the fact that GC is a heterogeneous disease. Specific HLA alleles may have the capability to modulate the presentation of peptides derived fromH. pyloriinfection to T cells. As a result of this presentation, the type or severity of T cell response may affect the proliferation of a lineage-specific malignant T cell clones[5].
In the study by Liet al[10], the HLA-CW*03 ratio was significantly higher in cases with increased risk of GC andH. pylori-infected patients. A variety of exogenous stimulations such as toxins ofH. pyloriwith gastric and bile juices always affect the gastric mucosa[40]. These stimulations cause epithelial cells to secrete IL-12 and induce natural killer (NK) cells. These NK cells secrete IFN-g, which initiates the Th1 immune response from naive T cells and causes phenotypic changes in epithelial cells, resulting in the upregulation of HLA-DR and HLA-B27 genes[41].
Different mechanisms may be responsible for the synergistic effect ofH. pyloriinfection and specific HLA genotypes. Specific HLA genotypes may influence the immune response and lead to carcinogenesis after the initial infection[17]. However, the common consensus is that genetically different host responses against the virulence factors ofH. pylorimay cause inflammation with varying intensities, as well as gastric epithelial erosions with different stages.
Genetic and epigenetic factors may also influence the severity of inflammation and thereby contribute to different clinical outcomes. Upregulation of significant MHC class II type genes in epithelial cells may be related to the activation of T cells in the lamina propria and macrophages, as well as the consequent release of cytokines such as interferon-γ. Confirming this hypothesis, upregulated expression of MHC class II genes in gastric epithelial cells had a positive correlation with the T cells in the lamina propria in children withH. pyloriinfections according to Lopeset al[42].
We could not detect any HLA allele as an independent risk factor for GC under our criterion, but without any criterion, some HLA alleles may regulate host susceptibility toH. pyloriinfection in the presence of its virulence factors. It can be suggested that some immunogenetic factors of the host may have important effects forH. pyloriinitiated inflammation leading to carcinogenesis. Although various host orH. pylorivirulence factors may have a role in the development of GC, specific host factors like HLAs could also modulate GC susceptibility or the resistance of individuals. Individuals who are at risk due to the susceptibility of HLA alleles to GC should be monitored prior to the initiation of carcinogenesis.
We only focused on the association between HLA gene polymorphisms and multiple EPIYA-C repeats ofH. pyloriDNAs isolated from the gastric biopsy specimens. Only two of our control group cases had multiple EPIYA-C repeats, so we compared two subgroups of the study group. We could not find a significant difference between GC and DU subgroups in terms of multiple EPIYA-C repeats. On the other hand, in a simple comparison between study and control groups, HLADQA1*01 (OR = 1.848), HLA-DQB1*06 (OR = 1.821), and HLA-A*2 (OR = 1.579) alleles were detected as independent risk factors for GC and DU.
This study has some limitations. The resolution of the HLA kits was low, which prevented an exact HLA comparison with other studies. We also did not evaluate some HLA alleles, such as HLA-C. Other important result of our study was that HLADR1*13-HLA-DQA1*01-HLA-DQB1*06 haplotype frequency was detected significantly higher in our GC subgroup cases more than both DU and control group cases. This results is similar for the locus type but different for HLA allele types from the study of Andoet al[43]They reported DRB1*04:05-DQA1*03:03-DQB1*04:01 haplotype frequency with 10%–30% in Japanese population and the risk of GC development.
CONCLUSION
This is the first study to evaluate the association between HLA alleles with GC and DU and asymptomaticH. pyloricases. Even though no HLA alleles were detected in multivariate analysis, HLA-DQB1*06 was significantly less frequent in the GC subgroup with multiple EPIYA repeats in the univariate analysis. However, this HLA allele was not detected as an independent risk factor in the multivariate analysis. In the multivariate logistic regression analysis using significantly different alleles and no discriminative criteria, HLA-DQA1*01 (OR = 1.848), HLA-DQB1*06 (OR = 1.821), and HLA-A*02 (OR = 1.579) alleles were found to be risk factors for GC and DU. However, we suggest that these HLA alleles make individuals prone to the development of GC without cagA and multiple EPIYA C repeats of the host. To clarify the effects of HLA alleles on the pathogenesis of gastric malignancies, more comprehensive, prospective, large-scale studies with high HLA resolution detection should be performed in the future.
ARTICLE HIGHLIGHTS

ACKNOWLEDGEMENTS
We thank Assoc. Prof. Dr. Murat Telli from the Department of Biology in Bolu Abant Izzet Baysal University, Turkey for the calculation of H-W equilirum and LD.
猜你喜欢
天津音乐学院学报(2022年1期)2022-05-23
销售与市场(营销版)(2020年6期)2020-06-20
中学生英语·阅读与写作(2017年10期)2017-11-14
食品安全导刊(2017年8期)2017-08-16
汽车博览(2016年10期)2016-11-11
读写算·教研版(2016年6期)2016-03-28
农产品市场周刊(2015年49期)2016-02-01
小雪花·成长指南(2014年4期)2014-05-26
中学英语之友·中(2008年9期)2008-10-18
 World Journal of Gastroenterology2020年32期
World Journal of Gastroenterology2020年32期
- World Journal of Gastroenterology的其它文章
- Features of extrahepatic metastasis after radiofrequency ablation for hepatocellular carcinoma
- Emergency department targeted screening for hepatitis C does not improve linkage to care
- Dual targeting of Polo-like kinase 1 and baculoviral inhibitor of apoptosis repeat-containing 5 in TP53-mutated hepatocellular carcinoma
- New advances in radiomics of gastrointestinal stromal tumors
- Current status of Helicobacter pylori eradication and risk factors for eradication failure
- Development of a novel score for the diagnosis of bacterial infection in patients with acute-on-chronic liver failure