
g-C3N4富集结合毛细管电泳与电感耦合等离子体质谱联用分析西瓜中硒形态
2020-09-18 09:21冯金素曹玉嫔莫桂春唐莉福邓必阳
色谱 2020年10期
冯金素, 曹玉嫔, 莫桂春, 唐莉福, 邓必阳
(药用资源化学与药物分子工程国家重点实验室, 广西师范大学化学与药学学院, 广西 桂林 541004)
硒元素是促进植物生长的有益元素,也是人体和动物必需的微量元素之一[1,2],具有增强机体免疫力、抗衰老、抑制毒性、抗癌等功效,且与人的甲状腺代谢有着密切的关系[3-6]。在自然环境中,硒元素主要以无机硒和有机硒两种形式存在。高等植物体中的无机硒含量相对较少,大部分以有机硒的形态存在。在生物体中,硒主要以硒蛋白等有机硒化合物的形态存在。自然环境中硒过量可能对水生生物产生各种不利影响[7],人体摄入量过多时会引起硒中毒,造成毛发易脱落、指甲易碎裂脱落、肠胃不适、秃头、皮肤红疹、倦怠、情绪不稳定、四肢无力发麻、肝脏损害等[8-10]。研究表明,许多疾病的发生与硒的摄入量偏低有关,如肿瘤、糖尿病、克山病、大骨节病、生殖功能障碍、高血压等[11-13]。硒的生物有效性以及毒性在很大程度上取决于硒的化学形态[1,14,15]。人体获取硒直接或间接源于动植物,但从日常食品中摄入硒不能完全满足人体的需要,需要从富硒食品中摄取,因此,对富硒产品进行研究具有现实意义。
硒元素在体内的适宜浓度范围很窄[16], 1998年,世界卫生组织(WHO)推荐:儿童每天摄入硒的量为6~21 μg,青少年为26~30 μg,成年人为26~35 μg,硒摄入的上限为400 μg。硒在实际样品中含量较低,因此对分析方法的灵敏度和准确度要求比较高。硒形态分析的研究方法和应用技术已有报道[17-23]。目前,硒形态分析的方法主要有高效液相色谱联用电感耦合等离子体质谱(ICP-MS)[24,25]、气相色谱联用ICP-MS[26]、液相色谱联用氢化物发生原子荧光光谱[27]、流动注射联用氢化物发生电感耦合等离子体原子发射光谱[28]、毛细管电泳(CE)联用电热原子吸收光谱[29]和CE-ICP-MS[30]等,但是大多数已报道的方法存在分析硒形态数目较少、样品用量较多等不足。因此,需要建立一种具有广泛应用前景、灵敏度更高的新方法以满足硒形态分析的需要。在众多分析方法中,CE具有分离效率高、样品消耗少、分析速度快等优点[31],而ICP-MS具有元素选择性好、线性范围宽和检出限低等优点,CE与ICP-MS联用可成为元素形态分析强有力的分析工具,有着广泛的应用前景[32-34]。本文建立了CE-ICP-MS用于西瓜中6种硒形态的分析,讨论了分析条件及类石墨烯氮化碳(g-C3N4)富集对硒形态分析的影响。该方法具有灵敏度高、分离速度快等特点。
1 实验部分
1.1 仪器
NexION300X型ICP-MS仪器配备一个动态反应池(Dynamic Reaction Cell, DRC)系统(PerkinElmer,美国); MD6C-6HL型微波样品处理系统(北京盈安美诚科学仪器有限公司); H1650-W型医用离心机(湖南湘仪实验室仪器开发有限公司); Spectrum Two FT-IR傅里叶变换红外光谱仪(Perkin-Elmer,美国); Rigaku D/max 2500/pc型X射线粉末衍射仪(XRD, Rigaku,日本);场发射环境扫描电子显微镜(Elementar,德国);熔融硅石英毛细管(长100 cm,内径100 μm、外径365 μm,河北永年锐沣色谱器件有限公司); HV-303 P1高压电泳电源(天津圣火科技有限公司);自组装毛细管电泳压力进样装置;SK3310HP超声波清洗器(上海科导仪器有限公司); DHG-9035A电热恒温鼓风干燥箱(上海启信科学仪器有限公司); W-D20型超纯水系统(北京盈安美诚科学仪器有限公司)。
CE-ICP接口按参考文献[35]制作,该接口外观类似同心玻璃雾化器,雾化器中心管被毛细管所代替,它的外端口长约0.5 cm,内径0.5 mm,样品出口端为负极。

表 1 毛细管电泳及电感耦合等离子体质谱工作条件
1.2 试剂
100 mg/L硒(Se)标准溶液(GSB G62029-90(3401), PerkinElmer,美国); SeO2(AR,北京市朝阳区中联化工试剂厂), Na2SeO4(AR,天津市化学试剂研究所);硒代蛋氨酸(DL-selenomethionine, SeMet) (纯度为99%, Alfa,美国);硒代胱氨酸(L-selenocystine, SeCys2) (纯度为98%,北京百灵威公司);硒脲(selenourea, SeUr) (纯度99%, Alfa,美国);硒代乙硫氨酸(selenoethionine, SeEt) (纯度为98%, TRC,加拿大);硼酸钠(Na2B4O7·10H2O)(天津市福晨化学试剂厂);磷酸二氢钠(NaH2PO4·2H2O)、30%过氧化氢、乙酸、甲酸、乙醇(AR,西陇化工股份有限公司);三聚氰胺和浓硝酸(AR,上海阿拉丁生化科技股份有限公司);十六烷基三甲基溴化铵(CTAB) (AR,湖南湘中化学试剂开发中心)。配制质量浓度均为1 g/L (按硒计)的Se(Ⅳ)、Se(Ⅵ)、SeMet、SeCys2、SeUr、SeEt标准溶液,于4 ℃保存,使用时逐级稀释到所需浓度。
所有试剂用前均未进一步纯化,实验用水均为超纯水。实验前所有溶液均经过0.45 μm水性滤膜过滤以除去粒径大的物质,防止堵塞毛细管,并经超声波清洗器超声7 min (功率为180 W,工作频率为53 kHz)以除去溶液中的气泡防止电泳过程中出现电流断流现象。
1.3 检测条件
新毛细管用0.1 mol/L NaOH溶液冲洗12 h,再用超纯水冲洗1 h。每次实验前先分别用0.1 mol/L NaOH、超纯水和运行缓冲液各冲洗10 min。进样后换上缓冲液加载一定的高压进行电泳分离,然后利用ICP-MS来检测分离峰。CE-ICP-MS的工作气体为氩气,电泳温度为22 ℃,实验条件见表1。
1.4 样品处理
1.4.1样品中总硒的提取
分别取普通西瓜、富硒西瓜的西瓜汁和西瓜渣各3份,每份准确称取0.200 0 g于微波消解罐中,加入5 mL浓HNO3和1 mL 30% H2O2,微波消解升温过程为:第一步升温至120 ℃并保持3 min,第二步升温至150 ℃并保持3 min,第三步升温至180 ℃并保持5 min, 3个步骤的升温速率均为8 ℃/min。消解完全后,冷却至室温,打开消解罐,将样品溶液转移至离心管中,用超纯水洗涤消解罐和盖子3次,合并洗涤液,用超纯水定容至20 mL,同样的方法制备空白溶液,备用。
1.4.2g-C3N4的合成及样品中硒形态的提取
称取2.0 g三聚氰胺于反应釜中,在600 ℃下密闭加热2 h,冷却至室温,取0.5 g上述产物置于烧杯中在室温下超声10 h,静置30 min,取上层乳白色溶液于60 ℃下烘干,即得黄色g-C3N4薄片。
称取0.500 0 g富硒西瓜汁和西瓜渣各3份于15 mL离心管中,再分别加入0.008 0 g胃蛋白酶和10 mL超纯水,置于超声波清洗器中在37 ℃水浴中超声2 h,然后以4 000 r/min转速离心20 min,重复提取2次,合并提取液并定容至40 mL。分别取10 mL上述富硒西瓜汁和西瓜渣样品各3份于15 mL离心管中,加入0.010 0 g g-C3N4,超声吸附8 min,然后以8 000 r/min转速离心20 min,弃去溶液。加入0.5 mL 1.0 mol/L NaOH溶液作为洗脱剂,超声8 min,收集上层清液,经0.45 μm水性滤膜过滤即得富硒西瓜样品溶液。同样方法制备普通西瓜样品溶液和空白溶液。
2 结果与讨论
2.1 硒干扰的消除
硒在自然界中有6种同位素,丰度最大的是80Se(49.61%),其次是78Se(23.77%)。然而,80Se和78Se在测定时分别受到40Ar40Ar+和38Ar40Ar+的干扰。由于CH4与40Ar40Ar+反应,可以消除40Ar40Ar+对80Se的干扰,因此,本实验采用CH4作为动态反应气[36],并对CH4的流速进行了优化。结果表明,随着动态反应气流速的增加、30 μg/L Se(Ⅳ)峰强度逐渐下降,当动态反应气流速增到1.1 mL/min之后,30 μg/L Se(Ⅳ)峰强度变化不大,此时干扰被消除,考虑到检测灵敏度,选择动态反应气流速为1.1 mL/min。
2.2 电泳电压和进样时间对分离效率的影响
电压是影响毛细管电泳分离的一个因素。在8 mmol/L NaH2PO4-12 mmol/L H3BO3-0.2 mmol/L CTAB (pH=9.2)的电泳缓冲液中,比较电泳电压在19~25 kV范围内对分离效率的影响。结果表明,电压的增大会改善样品的分离度并缩短迁移时间,但是电流也会随之增加、焦耳热效应明显。综合以上因素选择电泳电压为22 kV。在22 kV分离电压下,考察进样时间分别为9、10、11、12、13及14 s对硒形态峰的影响,发现进样时间在12 s以后峰高增幅不大,且较长的进样时间会使峰形的重现性变差、峰变宽,因此选择进样时间为12 s。
2.3 缓冲液浓度对分离效率的影响
毛细管电泳的分离效率与其缓冲液的浓度有着较大的关系,因为它会影响到样品的电泳淌度,从而影响毛细管电泳的分离效率。本实验在NaH2PO4-H3BO3(摩尔浓度比为2∶3, pH 9.2)中加入CTAB作为阳离子表面活性剂用以修饰毛细管表面。通过添加不同浓度的CTAB (0.1、0.2、0.3、0.4、0.5 mmol/L),考察CTAB浓度对6种硒形态分离的影响,结果表明,当CTAB浓度小于0.2 mmol/L时,CE电流不稳定,而当CTAB浓度高于0.3 mmol/L时,6种硒化合物的分离效率变差。考虑到分离效率和重复性,本实验选择0.2 mmol/L作为CTAB浓度。

图 1 缓冲液浓度对硒形态保留时间的影响Fig. 1 Effect of buffer solution concentration on retention time of selenium species a. 4 mmol/L NaH2PO4-6 mmol/L H3BO3-0.2 mmol/L CTAB; b. 6 mmol/L NaH2PO4-9 mmol/L H3BO3-0.2 mmol/L CTAB; c. 8 mmol/L NaH2PO4-12 mmol/L H3BO3-0.2 mmol/L CTAB; d. 10 mmol/L NaH2PO4-15 mmol/L H3BO3-0.2 mmol/L CTAB; e. 12 mmol/L NaH2PO4-18 mmol/L H3BO3-0.2 mmol/L CTAB. CSeUr, SeCys2, SeMet, Se(Ⅳ), Se(Ⅵ), SeEt=30 μg/L. For other conditions, see Table 1.
考察了不同浓度的NaH2PO4-H3BO3缓冲液对6种硒形态分离的影响,如图1所示,较高浓度的缓冲液使得迁移时间延长。考虑硒形态的峰形及分离度,本实验选择0.2 mmol/L CTAB-8 mmol/L NaH2PO4-12 mmol/L H3BO3(pH 9.2)作为运行缓冲液。
2.4 缓冲液pH值对分离效率的影响
缓冲液的pH值对分析物的分离同样有着较大的影响,确定了缓冲液的浓度后,本实验考察了pH 8.6~9.4范围内的缓冲液对6种硒形态保留时间的影响,结果见图2。由图2可知,随着pH值增大,SeMet、Se(Ⅵ)、SeEt的保留时间逐渐减少;当pH值为9.4时,只出现5个峰,SeCys2和SeMet已经重叠;当pH值为9.2时,6种硒形态分离效果最佳。因此,本实验选择缓冲液pH值为9.2。

图 2 缓冲液pH对6种硒形态保留时间的影响Fig. 2 Effect of the pH of buffer solution on retention time of selenium species CSeUr, SeCys2, SeMet, Se(Ⅳ), Se(Ⅵ), SeEt=30 μg/L. For other conditions, see Table 1.
2.5 标准溶液中SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt的形态分析
在优化的实验条件下(见表1),利用CE-ICP-MS测定质量浓度均为30 μg/L(按硒计)的SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt的混合标准溶液(见图3)。为了确定各个硒形态的出峰位置,采用在混合标准溶液中加入单标定位的方法。在30 μg/L硒混合标准溶液中分别加入30 μg/L SeUr、30 μg/L SeCys2、30 μg/L SeMet、30 μg/L Se(Ⅳ)、30 μg/L Se(Ⅵ)、30 μg/L SeEt,然后分别利用CE-ICP-MS进行测定。通过与图3对比可知,在30 μg/L硒混合标准溶液中加入单标硒形态后,加入的单标硒形态峰明显比图3中对应硒形态峰高,而其他硒形态峰的迁移时间和峰强度几乎不变,由此可确定6种硒形态出峰顺序依次为SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)、SeEt,其保留时间分别为2.567、3.051、4.401、7.286、8.166、10.126 min。

图 3 6种硒形态的CE-ICP-MS图Fig. 3 Electropherogram of six selenium species using CE-ICP-MS CSeUr, SeCys2, SeMet, Se(Ⅳ), Se(Ⅵ), SeEt=30 μg/L. For other conditions, see Table 1.
2.6 g-C3N4的表征
根据相关报道可知[36], g-C3N4的XRD谱图在2θ角为27.4°和13.1°处有特征峰。本实验对所制备的产品进行XRD检测,其XRD谱图出现了g-C3N4的特征峰,而没有其他杂质峰出现,表明所制备的产品具有g-C3N4的结构组成。两个特征峰中,以27.4°的特征峰强度最强,其归属于g-C3N4的(002)晶面,是g-C3N4芳香环系统的层间堆垛峰,其晶格间距(d, 0.326 nm)与g-C3N4的层间距对应。而2θ角为13.1°处的特征峰归属于g-C3N4的(100)晶面,与组成g-C3N4的Melem(CN环)单元形成的层内空间距离相对应[37]。为了进一步确定g-C3N4样品的组成,采用FT-IR对样品进行了表征,在800 cm-1、1 200~1 600 cm-1和2 800~3 400 cm-1处出现了吸收带,与g-C3N4经典的FT-IR谱图一致,这说明g-C3N4已经成功合成。其中在800 cm-1的吸收峰是组成g-C3N4的单元三嗪环(triazine)的碳氮环的弯曲振动特征峰;1 200~1 600 cm-1的吸收带则是g-C3N4的碳氮杂环上的C=N、C-N和环外C-N伸缩振动吸收峰;而在2 800~3 400 cm-1的吸收带则可能是g-C3N4边缘破损芳香环上的-NH和-NH2基团的伸缩振动,或是其表面上吸附的水分子的伸缩振动[38]。
2.7 萃取条件的优化
2.7.1g-C3N4吸附率的优化
固定10 mg g-C3N4量不变,分别对10 mL 5~90 μg/L的SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt混合标准溶液进行测定,通过公式(1)计算吸附率(T),结果表明:10 mg g-C3N4对体积为10 mL的20 μg/L SeUr、50 μg/L SeCys2、40 μg/L SeMet、60 μg/L Se(Ⅳ)、75 μg/L Se(Ⅵ)和30 μg/L SeEt可完全吸附(见图4)。
(1)
其中,C0为初始质量浓度,C为g-C3N4溶液中剩余的质量浓度,单位为μg/L。

图 4 类石墨烯氮化碳对硒形态的吸附率Fig. 4 Adsorption percentages of selenium species using g-C3N4For detection conditions, see Table 1.
2.7.2洗脱剂的选择
本实验考察了NaOH洗脱剂对6种硒形态洗脱效果的影响(见图5)。固定NaOH体积0.5 mL,当NaOH浓度为1.0 mol/L时,6种硒形态均可回收完全,因此选择NaOH溶液的浓度为1.0 mol/L。

图 5 NaOH溶液浓度对硒形态洗脱的影响Fig. 5 Effect of NaOH concentration on the desorption of selenium species Mass concentration: SeUr 20 μg/L, SeCys2 50 μg/L, SeMet 40 μg/L, Se(Ⅳ) 60 μg/L, Se(Ⅵ) 75 μg/L, SeEt 30 μg/L . For detection conditions, see Table 1.
对1.0 mol/L NaOH溶液体积进行了优化,结果表明6种硒形态从g-C3N4上完全洗脱的体积为0.5 mL,因此选择0.5 mL作为洗脱体积。
2.7.3超声时间的影响
超声时间的优化既可保证吸附洗脱完全,又关系到实验的速度快慢。在1~10 min内考察超声时间对SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt吸附和洗脱效果(见图6),得到最佳吸附时间为8 min,最佳洗脱时间为8 min。
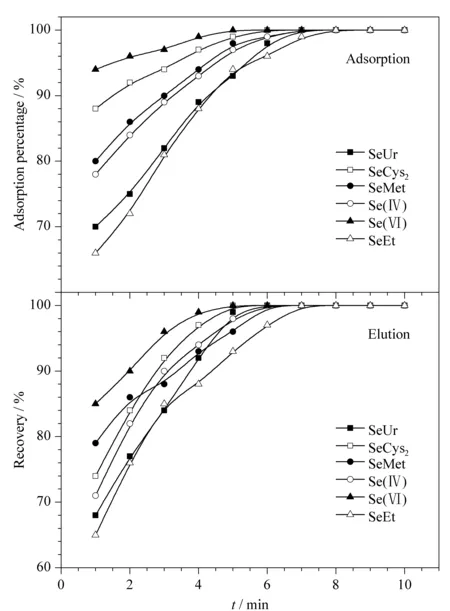
图 6 超声时间对硒形态吸附和洗脱的影响Fig. 6 Effect of ultrasonic time on adsorption and elution of selenium species CSeUr, SeCys2, SeMet, Se(Ⅳ), Se(Ⅵ), SeEt=20 μg/L. For detection conditions, see Table 1.
2.8 分析性能
在优化的实验条件下考察本方法的分析性能,SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt的检出限分别为6.2、30、11、8.2、48和5.5 ng/L(按硒计,计算测量11次空白溶液信号的标准偏差的3倍所对应的质量浓度),比相关研究[18,23,24]的检出限低(如表2)。上述硒形态的相对标准偏差(RSD,n=5)在2.2%~3.5%之间,相关系数>0.999 5。线性范围、富集因子(EF, EF=富集后校正曲线斜率/富集前校正曲线斜率)及相关文献报道的检出限见表2。

表 2 用CE-ICP-MS检测硒形态的分析性能(n=5)
2.9 提取剂的选择
理想的预处理方法既要将各种硒形态从待测物中高效地提取出来,还要尽量避免各种硒形态在处理过程中发生变化。由于所测定的6种硒形态都具有较好的水溶性,因此不采用有机溶剂作为提取剂。本实验分别采用超纯水、0.1 mol/L HCl、胃蛋白酶作为提取剂分别提取西瓜汁和西瓜渣样品中的硒形态,以提取剂对硒形态的提取量为依据选择本实验的提取剂。根据已报道文献[39],大多数硒形态的提取需要时间较长,本实验采用超声波辅助提取,较大地缩短了提取时间,再经g-C3N4进行富集显著提高灵敏度。实验结果表明,使用200 mg/L胃蛋白酶作为提取剂,西瓜汁中硒形态含量比使用0.1 mol/L盐酸作为提取剂所得硒形态含量高出80%以上;西瓜渣中硒形态含量比使用0.1 mol/L盐酸作为提取剂所得硒形态含量高出85%以上,因此,本实验选择胃蛋白酶作为提取剂。
2.10 样品分析
在优化的实验条件下用CE-ICP-MS联用技术分别测定富硒西瓜和普通西瓜样品中的硒形态。图7A(a)、7A(b)分别为用该方法测定普通西瓜汁和富硒西瓜汁的电泳图,图7A(c)为在富硒西瓜汁样品中加入10 μg/L SeCys2的电泳图。从图7A(a)中可知,所测普通西瓜汁中不含硒形态。对比图3与图7A(b)和图7A(c)可知,富硒西瓜汁样品中含有SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)和SeEt 5种硒形态。图7B(a)、图7B(b)分别为普通西瓜渣和富硒西瓜渣的电泳图;图7B(c)为在富硒西瓜渣样品中加入10 μg/L SeCys2的电泳图,从图中可知,图7B(c)中的峰1明显比图7B(b)中的峰1高,而其他硒形态峰的峰高及迁移时间几乎不变。对比图3与图7B(b)和图7B(c)可知,所检测富硒西瓜渣中含有SeCys2、SeMet和SeEt 3种硒形态。

图 7 (A)西瓜汁和(B)西瓜渣中硒形态的CE-ICP-MS图Fig. 7 Electropherograms of selenium species in (A) watermelon juice and (B) watermelon slag using CE-ICP-MS a. ordinary watermelon; b. selenium-rich watermelon; c. selenium-rich watermelon+10 μg/L SeCys2. For detection conditions, see Table 1.
采用微波消解法同时处理富硒西瓜和普通西瓜样品,用ICP-MS测得富硒西瓜汁与富硒西瓜渣样品中硒总含量分别为78.0 ng/g和27.7 ng/g,然而在普通西瓜样品中没有检测到硒。用CE-ICP-MS测定富硒西瓜汁和富硒西瓜渣样品中硒总量分别占ICP-MS测定富硒西瓜汁与富硒西瓜渣样品中硒总量的96.8%和97.5%,富硒西瓜渣中硒形态为有机硒形态,富硒西瓜汁中既包含无机硒形态也包含有机硒形态,所测结果见表3。加标回收率在96.0%~106%之间。
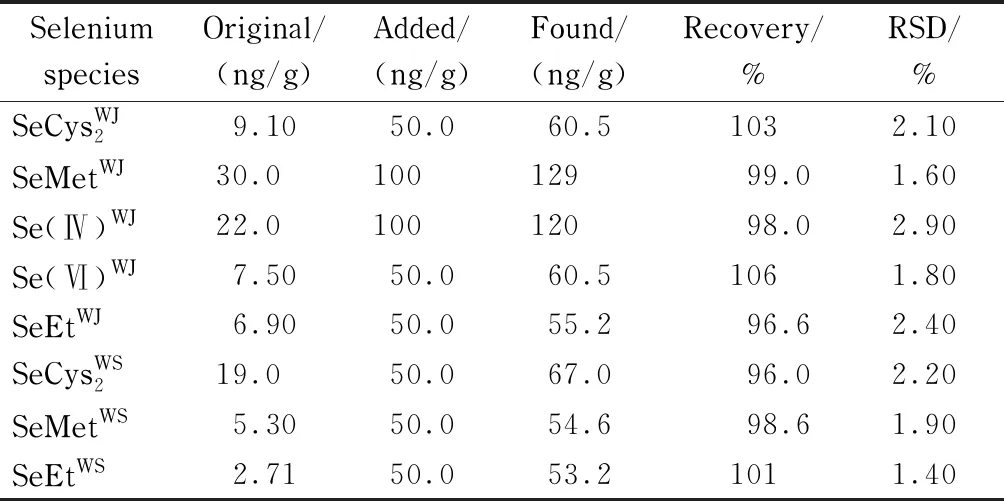
表 3 富硒西瓜样品中硒形态测定结果及回收率(n=5)
3 结论
本文建立了CE-ICP-MS联用技术用于西瓜中硒形态分析的新方法,在11 min内可将SeUr、SeCys2、SeMet、Se(Ⅳ)、Se(Ⅵ)、SeEt 6种硒形态良好分离。该方法也可应用于其他食品及环境中硒形态的分析。
猜你喜欢
东方少年·阅读与作文(2021年4期)2021-06-10
园艺与种苗(2021年4期)2021-05-26
食品与机械(2021年4期)2021-05-10
西部资源(2020年6期)2020-03-01
故事作文·高年级(2018年11期)2018-11-19
意林(2018年3期)2018-03-02
食品安全导刊(2017年36期)2017-02-01
中国粮油学报(2016年1期)2016-02-06
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
