
三种联结方式对典型双四唑含能材料晶体结构和性能的影响
2020-09-17 08:49:38林秋汉王鹏程
含能材料 2020年9期
林秋汉,李 鑫,王鹏程,陆 明
(南京理工大学化工学院,江苏 南京 210094)
1 引言
四唑环中氮含量超过80.0%,是除全氮阴离子(N5-)之外氮含量最高且能常温稳定存在的结构单元。四唑化合物特别是双环和多环四唑衍生物,因其结构中含有大量N─N 键、C─N 键等高势能键以及大的环张力,并且具有高密度、高生成焓、热稳定性好等一系列优良性质,而成为理想的含能化合物。此外,以不同联结方式合成得到的双环四唑比单环四唑具有更多的修饰位点,可以通过不同取代基团的引入更好地调节性能。根据连接方式的不同,双环四唑化合物可以分为偶氮四唑、联四唑、联氨四唑等(如图1)[1-5],双环四唑类化合物因其氮含量高、结构致密以及燃烧或爆炸的最终产物主要为N2等特点,可以避免环境污染和健康危害,作为潜在的含能材料在混合炸药、低特征信号推进剂等方面进行的研究引起了人们极大兴趣[6]。
2012 年,德国慕尼黑大学的Fischer 等[7]报道合成了一种双四唑含能离子盐5,5″‑联四唑‑1,1′‑二氧羟铵盐(TKX‑50)。TKX−50 不仅具有较高的能量水平,且感度较低,是一种很有应用价值的高能低感单质炸药,随后,国内外相继掀起了一场研究TKX‑50 的热潮。西安近代化学研究所[8]、北京理工大学[9]、南京理工大学[10]、中国工程物理研究院[11-12]、中北大学[13]等高校及科研院所都参与其中,对TKX‑50 的合成、理论计算及应用研究进行了一系列的探索,并取得了一系列进展。TKX‑50 通过联四唑提升性能,其理论爆速达到9698 m·s-1,爆压为42.4 GPa。TKX‑50 的分子结构中没有─NO2等致爆基团,但仍具有较高的爆轰性能,被认为是释能方式有别于传统含能材料分子内氧化还原反应的新一代含能材料。根据单晶结构[7,14-15]还发现,在氮环上引入氧原子,不仅与阳离子中的羟氨构成了分子内的氢键体系,使其密度达到了1.918 g·cm-3,而且降低了感度。
偶氮键(─N=N─)是含能材料的一个重要基团,常被引入到富氮化合物中得到富氮双环化合物,不但可以提高氮含量,且可以大幅度增加体系生成热,同时也丰富了富氮化合物的研究内容,成为富氮杂环化合物研究的一个重要发展方向,在TKX‑50 分子结构中引入偶氮键(─N=N ─)得到5,5′‑偶氮四唑‑1,1′‑氮氧化物二羟氨盐(ATZO‑1)[14],更高氮含量的ATZO‑1 是否具有更为优越的性能值得期待;而在偶氮四唑上引入氧化偶氮键(─N=N(O)─)得 到5,5′‑偶 氮 四 唑‑5‑氮 氧 化物[15]与TKX‑50 分子亦有极为相似的结构。双四唑化合物作为一类重要的含能化合物,现如今已发展了很多不同联结方式的双四唑类含能化合物,如C桥连、N 桥连、偶氮键联结、氧化偶氮键联结等,本研究结合晶体结构与量化计算数据探究偶氮键和氧化偶氮键联结的双四唑化合物是否和TKX‑50 具有类似的特点,根据单晶数据探讨了这三种相似化合物晶体结构的微观区别,并采用玻恩‑哈伯循环(Born−Haber cycle)和热力学中赫斯(Hess)定律计算了它们的生成热,根据K‑J 方程计算得到了其爆轰参数,总结了偶氮键(─N=N ─)和氧化偶氮键(─N=N(O)─)的引入对双四唑类含能材料能量及安全性能的影响。
2 计算方法
不同文献针对含能材料理化性能的预测采用不同的计算方法,因此其计算数值可能会有一定的差距。为了更为科学地探讨(─N=N─和─N=N(O)─键的引入对双四唑类含能材料性能的影响,本研究参考文 献[7,14]的 单 晶 密 度,统 一 采 用 玻 恩‑哈 伯 循 环(Born‑Haber cycle)和热力学中赫斯(Hess)定律计算了相关化合物的生成热,根据Kamlet‑Jacobs 公式(K‑J方程)重新计算了其爆轰性能数据,在此基础上进行比对分析。由于5,5′‑偶氮四唑‑5‑氮氧化物的二羟氨盐(ATO‑1)目前未被报道,其密度和生成热均采用量化计算方法获得。
2.1 Gaussian 软件计算
根据玻恩‑哈伯循环(Born‑Haber cycle)和热力学中赫斯(Hess)定律[16],离子化合物固相标准生成焓(ΔHfθ)、晶格能(ΔHL)和晶格势能(UPOT)可按式(1)计算:

式中,ΔHL代表离子化合物的晶格能,kJ·mol-1。从式(1)可见,晶格能是预测离子化合物生成热所比不可少的一个重要参数,晶格能可以通过詹金斯公式[16]求得:
式中,nM和nX分别决定于Mp+阳离子和Xq−阴离子。当离子为单原子离子时,n=3;当离子为线性多原子离子时,n=5;当离子为非线性多原子离子时,n=6。而式UPOT代表晶格势,可由式(3)计算:

式中,ρm代表离子化合物的密度,g·cm-3;Mm代表离子化合物的分子量。参数γ(kJ·mol-1·cm)和δ(kJ·mol-1)参考文献[16]。
2.2 爆轰性能计算
对于分子式为CaHbOcNd的含能材料,采用传统的Kamlet‑Jacobs 公式(K‑J 方程)对所涉及化合物的爆轰参数进行计算[17-19]。
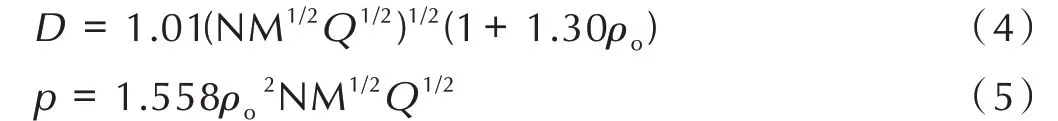
式中,D为爆速,km·s-1;p为爆压,GPa;ρo为化合物的密度,g·cm-3;N 为每克炸药爆轰生成气体产物的量,mol·g-1;M 为气体产物的摩尔质量,g·mol-1;Q为每克炸药的爆轰化学能,即单位质量炸药的最大爆热(J·g-1)。
TKX‑50 和ATZO‑1 的分子式中,c=1/2b,故其最大爆热Q值根据式(6)计算,而ATO‑1 的c<1/2b,故其最大爆热Q值根据式(7)计算:
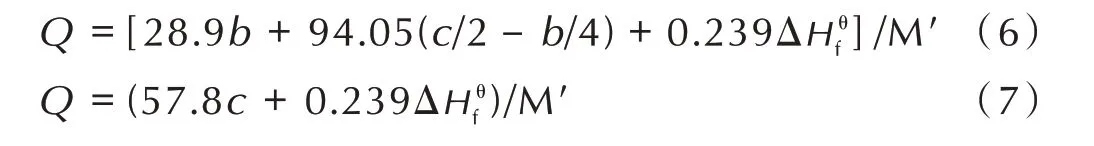
式中,M′是炸药的摩尔质量,g·mol-1。
3 ─N=N─和─N=N(O)─键对晶体结构的影响
3.1 TKX⁃50、ATZO⁃1 和ATO⁃Na 的晶体结构
ATZO‑1 与TKX‑50 结构相比较,其分子中仅多了一个─N=N─,它们的阳离子相同,ATO‑Na 的阳离子 为 金 属 离 子Na+;化 合 物TKX‑50、ATZO‑1 和ATO‑Na 的单晶结构如图1 所示,图1 显示,TKX‑50 和ATZO‑1 分子内阴阳离子间有不同强度的氢键作用,而ATO2-与Na+的主要存在离子键作用。

图1 化合物TKX‑50、ATZO‑1 和ATO‑Na 的单晶结构Fig.1 Crystal structures of TKX‑50,ATZO‑1,and ATO‑Na
3.2 ─N=N─和─N=N(O)─键对双四唑化合物的结构影响分析
化合物TKX‑50、ATZO‑1 和ATO‑Na 晶体中的部分键长值如表1 所示。在TKX‑50 的分子中,四唑环内的C—N 键长分别为0.1336 nm 和0.1332 nm,N─N键长在0.1306 ~0.1351 nm 之间,N─N 键键长区别较大;在ATZO‑1 的分子中,四唑环内的两个C—N 键长分别为0.134 nm 和0.1342 nm,键长略长于TKX‑50 分子的C—N 键;ATZO‑1 的分子内四唑环的N─N 键长为0.1328~0.1336 nm,键长基本接近,说明其四唑环的离域程度更高,环的稳定性优于TKX‑50;二者四唑环内的C—N 键均短于正常C—单键的键长(0.1470 nm),且长于C=N 双键的键长(0.1270 nm),说明四环内含有的C=N 双键与环内氮的孤对电子产生p‑π 共轭,键长趋于平均化。TKX‑50的分子中的N ─O 键键长分别为0.1317 nm 和0.1408 nm;而ATZO‑1 分子中的两个N‑O 键的键长分别为0.1306 和1.414 nm,长度差距大于TKX‑50,说明TKX‑50 分子的对称性要高于ATZO‑1 分子。

表1 化合物TKX‑50、ATZO‑1 和ATO‑Na 晶体中的部分键长比较Table 1 Comparison of selected bond length for TKX‑50,ATZO‑1,and ATO‑Na(Å)
而ATO‑Na 分子中,四唑环内C—N 键长分别为0.1333 nm 和0.1325 nm,N─N 键长在0.1314 nm~0.1339 nm 之间,也具有较好的芳香性,但是偶氮键上的氮氧键N→O 的键长为0.271 nm,远小于TKX‑50和ATZO‑1,说明偶氮键上的氧原子与偶氮键形成一定程度的共轭,与芳环上的N─O 键有一定程度的区别。
化合物TKX‑50、ATZO‑1 和ATO‑Na 的部分键角数据如表2 所示。可见,在TKX‑50 的分子中,四唑环内的键角介于105.49(12)~110.94(12)°之间,与正常键角(108°)最大偏差为2.94°,键角间最大偏差为5.45°;ATZO‑1 分子中的四唑环内键角介于104.81(16)°~112.16(16)°之间,键角间最大偏差为7.35°;ATO‑Na分子中的四唑环内键角介于102.27(5)°~114.44(17)°之间。键角同样趋于平均化,这是由于五元环内形成的大π 键导致的,芳香性的形成有助于其结构的稳定。TKX‑50 的分子中,联四唑环的键角N(1)─C(1)─C(1)为124.05(18)°,而ATZO‑1 内四唑环与偶氮键的键角N(1)─C(1)─N(5)为121.91(18)°,说明TKX‑50 的分子更为紧凑,而ATZO‑1 分子的拉伸程度更高,ATO‑Na分子四唑环与偶氮键的键角N(1)─C(1)─N(5)为126.54(18)°,拉伸程度最大。
此外,TKX‑50 分子中的二面角数据在180°或0°附近,例如N(2)─N(1)─C(1)─C(1)179.490(133)°;N(1)─C(1)─C(1)─N(4)0.128(247)°;N(1)─C(1)─C(1)─N(1)180.0(139)°;由此可见,N(1)与环平面最大偏角为-178.8°,即TKX‑50 分子内的两个四唑环基本在一个平面上。在ATZO‑1 分子中,二面角N(1)─C(1)─N(5)─N(5)为174.036(185)°;N(5)─C(1)─N(1)─N(2)为177.755(177)°;与TKX‑50 分子相比,ATZO‑1 分子在两个四唑环中间多了一个偶氮键(─N=N─)后,根据二面角数据可以看出,引入偶氮键(─N=N─)后,两个四唑环也几乎在同一平面上,但是发生一定程度的扭曲。同样,ATO‑Na 分子中两唑环的二面角与环平面最大偏角为-178.8°,亦发生轻微扭曲。由此可见,两个四唑环直接相连后两个四唑环基本共面,而以─N=N─和─N=N(O)─键连接则改变了其完美的平面结构。
根据晶包堆积图数据发现,TKX‑50 的晶胞结构中存在着大量的分子间氢键,且氢键作用很强。如O(2)─H(2)…N(1)间距为2.404 Å,N(5)─H(5c)…N(1)间距为2.670 Å,N(5)─H(5a)…O(1)间距为2.178 Å;O(2)─H(2)…N(1)的键角为144.543°,N(5)─H(5c)…N(1)的键角为162.842°,N(5)─H(5a)…O(1)的键角为145.962°;N(5)─H(2)…O(1)间形成离子键,其间距为1.699 Å,N(5)─H(2)…O(1)离子键的键角为140.430°,这是由于阳离子存在大量的─NH和─OH 等氢原子,极易与联四唑上的O、H 形成强的氢键作用。
化合物TKX‑50、ATZO‑1 和ATO‑Na 的部分氢键数据如表3 所示,由于阳离子的羟氨中H 的存在,ATZO‑1 的分子间亦存在大量的氢键,氢键作用力亦较强。如O(2)─H(2)…O(1)间距为1.994 Å,键角为158.43°;N(6)─H(6c)…N(4)间距为2.157 Å,键角 为176.463°;N(5)─H(2)…O(2)的 间 距 为2.851 Å,键角为129.739°。ATO‑Na 的结构中由于钠离子和结晶水的存在。也存在较强的分子内氢键和离子 键 的 相 互 作 用。 如O(3)─H(3B)…Na(1)(2.779 Å)形成分子内氢键,O(3)─H(3a)…Na(2)间距为2.8487(276)Å,O(4)─H(4a)…Na(1)间距为2.6479(219)Å,O(5)─H(5b)…Na(1)的键角为58.747(1984)°;N(6)─O(1′)…Na(2)形成离子键,N(1)…Na(2)间距为2.5718(17)Å,N(2)─Na(1)间距为2.4436(17)Å,N(2)─N(1)…Na(2)的键角为133.862(106)°;分子内和分子间较强氢键的存在是使这些化合物具有较高的密度和较好稳定性的原因之一(图2)。

表2 化合物TKX‑50、ATZO‑1 和ATO‑Na 晶体中的部分键角数据Table 2 Selected bond angles for TKX‑50,ATZO‑1,and ATO‑Na (°)
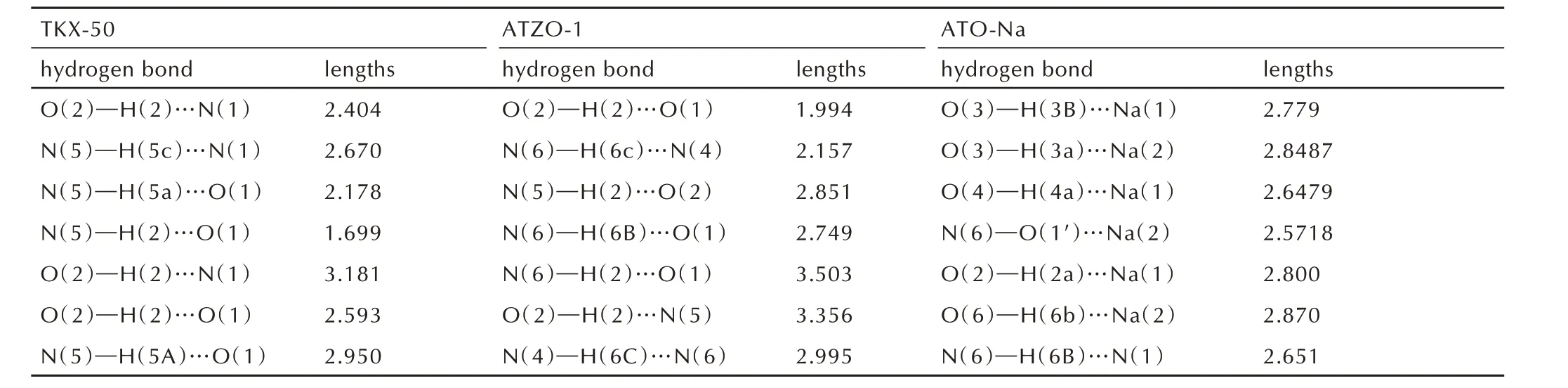
表3 化合物TKX‑50、ATZO‑1 和ATO‑Na 晶体中的部分氢键数据Table 3 Selected hydrogen bond length for TKX‑50,ATZO‑1,and ATO‑Na Å

图2 化合物TKX‑50、ATZO‑1 晶体结构中存在大量的分子间氢键Fig.2 A large number of intermolecular hydrogen bonds ob‑served in crystal packing of TKX‑50 and ATZO‑1
4 ─N=N─和─N=N(O)─键对双四唑化合物理化性能的影响
4.1 不同联结方式对双四唑化合物的密度和生成热影响
从表3 可以看出,以不同方式联结的三个化合物中,TKX‑50 的 密 度 最 高,达 到 了1.918 g·cm-3。而ATZO‑1 的 密 度 为1.778 g·cm-3,由 此 可 见,偶 氮 键(─N=N─)的引入不仅没有增加化合物的密度,反而使其密度大为降低。这可能是由于TKX‑50 中的两个四唑环更为紧凑,更有利于与阳离子NH3OH+形成更为紧密的氢键。根据晶体结构分析发现,ATZO‑1 和ATO‑Na由于两个四唑环以─N=N─和─N=N(O)─联结,使其结构有所拉伸,远不如TKX‑50 的结构致密,因此他们的密度均低于TKX‑50。ATZO‑1 和ATO‑1的氮含量分别为63.63%和67.74%,均高于TKX‑50(59.3%);偶氮键(─N=N─)的引入对于富氧平衡炸药来讲,亦能在一定程度上提高其氧平衡,由TKX‑50的-27.1 提高到了ATZO‑1 的‑24.22。
值得注意的是,ATZO‑1的生成热为721.8 kJ·mol-1,相比于TKX‑50的432.6 kJ·mol-1提高了289.2 kJ·mol-1,说明偶氮键的引入能够大大提高化合物的生成热。常见含能离子盐阴离子配体,一般为NO3-,ClO4-或者具有─NO2,─N─NO2等基团的多氮化合物,其生成热一般为-400~0 kJ·mol-1。由此可见,以本研究中提到的三个阴离子作为含能离子化合物的离子配体,其生成焓均具有明显的优势。 因此,引入偶氮键(─N=N─)或氧化偶氮键(─N=N(O)─)仍然为含能化合物分子设计的一个重要方法。
4.2 爆轰性能分析
根据单晶密度和理论计算求得生成热数据,再根据K‑J 方程估算得到的TKX‑50、ATZO‑1 和ATO‑1 的爆速爆压如表4 所示。

表4 TKX‑50、ATZO‑1、ATO‑1 和一些常见高能化合物的爆轰性能Table 4 Properties of TKX‑50,ATZO‑1,ATO‑1 and traditional high energy density materials
由于堆积效应的影响[7],TKX‑50 具有的高密度的优势使其具有最高的爆速和爆压数值(D=9314 m·s-1,p=39.94 GPa)。偶氮键(─N=N─)或氧化偶氮键(─N=N(O)─)联结的双四唑比直接相连的双四唑化合物具有更高的生成热,但是直接相连的双四唑化合物具有最高的生成热。ATZO‑1(D=9009 m·s-1,p=35.74 GPa)和ATO‑1(D=8823 m·s-1,p=34.05 GPa)的爆轰性能低于TKX‑50,因此同样以羟氨盐为阳离子配体,联四唑‑1,1′‑二氧化物阴离子的爆轰性能最具优势。从表3 可以看到,TKX‑50 的爆轰性能与HMX(D=9144 m·s-1,p=39.2 GPa)相当,ATO‑1 的爆轰性能 与 RDX(D=8830 m·s-1,p=35.9 GPa)相 当,ATZO‑1 则介于两者之间。
5 结论
研究了三种基于不同联结方式的双四唑环结构含能离子盐的晶体结构和理化性能.
(1)TKX‑50 的双四唑环结构更为紧密,且其分子内具有强的氢键作用,因此,其分子具有最高的晶体密度和爆轰性能,双四唑环以偶氮键(─N=N─)和氧化偶氮键(─N=N(O)─)拉伸了化合物的结构,使其密度有所降低。
(2)偶氮键(─N=N─)和氧化偶氮键(─N=N(O)─)联结的双四唑化合物在一定程度上增加了联四唑化合物的生成热。
(3)由于具有致密的结构和较高的密度,双四唑环直接相连的化合物TKX‑50 具最高的爆速和爆压数据(D=9314 m·s-1,p=39.94 GPa)。
(4)以 偶 氮 键(─N = N ─)和 氧 化 偶 氮 键(─N=N(O)─)联结的双四唑阴离子ATZO‑和ATO‑具有较高的氮含量和高生成热数据,以其作为含能离子盐的阴离子配体还是具有相当大的优势,可以作为设计新型的含能离子盐的阴离子配体候选物。
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
高中数理化(2022年16期)2022-09-14 13:57:06
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:18
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:38
中国粮油学报(2016年1期)2016-02-06 02:17:09
化工进展(2015年6期)2015-11-13 00:26:50
中国塑料(2015年11期)2015-10-14 01:14:05
火炸药学报(2014年5期)2014-03-20 13:17:51
火炸药学报(2014年5期)2014-03-20 13:17:46
