
蜜蜂球囊菌菌丝和孢子中微小RNA及其靶mRNA的比较分析
2020-09-12 10:16:52陈华枝祝智威蒋海宾王杰范元婵范小雪万洁琦卢家轩熊翠玲郑燕珍付中民陈大福郭睿
中国农业科学 2020年17期
陈华枝,祝智威,蒋海宾,王杰,范元婵,范小雪,万洁琦,卢家轩,熊翠玲,郑燕珍,付中民,陈大福,郭睿
蜜蜂球囊菌菌丝和孢子中微小RNA及其靶mRNA的比较分析
陈华枝,祝智威,蒋海宾,王杰,范元婵,范小雪,万洁琦,卢家轩,熊翠玲,郑燕珍,付中民,陈大福,郭睿
(福建农林大学动物科学学院(蜂学学院),福州 350002)
【】蜜蜂球囊菌(,球囊菌)专性侵染蜜蜂幼虫而导致白垩病。本研究旨在通过small RNA-seq(sRNA-seq)技术和生物信息学方法对球囊菌纯化菌丝(AaM)和纯化孢子(AaS)进行深度测序和比较分析,明确球囊菌菌丝miRNA和孢子miRNA的数量、结构和表达谱差异,并揭示菌丝和孢子共有miRNA、特有miRNA和差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其靶mRNA与球囊菌菌丝和孢子生长、发育和病原致病性的潜在关系。实验室条件下获得纯培养的球囊菌,利用sRNA-seq技术对AaM和AaS分别进行测序,通过对原始读段(raw reads)进行过滤和质控获得有效标签序列(clean tags)。通过Venn分析筛选菌丝和孢子共有miRNA和特有miRNA。根据≤0.05且|log2fold change|≥1的标准筛选AaM vs AaS的DEmiRNA。对上述共有miRNA、特有miRNA和DEmiRNA的靶mRNA进行预测,并对靶mRNA进行GO及KEGG数据库注释。根据靶向结合关系构建DEmiRNA和靶mRNA的调控网络。利用RT-qPCR验证测序数据的可靠性。AaM和AaS中分别得到12 982 320和12 708 832条raw reads,经过滤和质控分别得到10 800 101和9 888 848条clean tags。AaM中miRNA的长度介于18—26 nt,AaS中miRNA的长度介于18—24 nt,分布miRNA数量最多的长度均为18 nt,AaM和AaS中首位碱基为U的miRNA数量最多。AaM和AaS中表达量最高的miRNA均为miR6478-x、miR10516-x和miR482-x。菌丝和孢子共有miRNA靶向结合5 946个mRNA,二者特有miRNA分别靶向结合6 141和6 346个mRNA。共有miRNA的靶mRNA主要参与代谢进程、细胞进程和催化活性等42个功能条目,以及翻译、碳水化合物代谢和能量代谢等120条通路。AaM vs AaS比较组包含93个DEmiRNA,可靶向结合6 090个mRNA,这些靶mRNA可注释到38个功能条目和120条通路。DEmiRNA与靶mRNA之间形成较为复杂的调控网络,miR-4968-y位于调控网络的中心且能够靶向结合多达118个mRNA。RT-qPCR结果显示5个DEmiRNA的表达趋势与测序数据一致,证实了本研究中测序数据的可靠性。球囊菌菌丝和孢子中的miRNA具有类似的结构特征,但表达谱表现出明显差异;菌丝和孢子可能通过特异性表达和差异表达部分miRNA对其生长、发育和生殖进行调控。
蜜蜂球囊菌;菌丝;孢子;微小RNA;信使RNA;靶向结合
0 引言
【研究意义】蜜蜂球囊菌(,球囊菌)是一种特异性侵染蜜蜂幼虫的致死性真菌病原,能够使蜜蜂幼虫罹患白垩病,可导致成年蜜蜂数量和群势的急剧下降[1]。近期的研究表明,球囊菌对中华蜜蜂(,中蜂)幼虫和成虫、成年熊蜂也具有侵染性[2]。食物连同球囊菌孢子被蜜蜂幼虫摄入后进入中肠,幼虫期中肠与后肠之间存在一层隔膜,至预蛹期隔膜消失,孢子随食物残渣涌入后肠,在O2的刺激下孢子剧烈萌发、菌丝大量生长,先穿透肠壁再从尾部穿透体壁,进而包裹整个虫尸[3]。前人在球囊菌的分类鉴定[4-5]、增殖方式[6]和病理学[7-8]等方面开展了较多研究,但其分子生物学及组学研究较少。球囊菌微小RNA(microRNA,miRNA)等非编码RNA的相关研究尤为滞后。利用small RNA-seq(sRNA-seq)技术和生物信息学方法对球囊菌菌丝和孢子分别进行深度测序和组学分析,明确二者miRNA的数量和结构特征差异,揭示miRNA与菌丝生长、孢子萌发、杂交产孢、毒力因子合成与分泌等生物学过程的关系,可为明晰相关分子机理提供参考信息和数据基础。【前人研究进展】MiRNA是一类长度约为23 nt的细胞内源性小RNA分子,在真核生物中广泛存在且高度保守[9],可通过切割靶mRNA或阻止蛋白质的翻译过程来调控基因表达[10],从而参与调控生长发育及信号转导等相关进程[11]。自从在秀丽隐杆线虫()中发现第一个miRNA以来[12],人们已在动物[13]、植物[14]和微生物中[15-16]陆续发现了大量miRNA,但真菌miRNA的相关研究总体仍较为滞后。前期研究中,笔者团队已在mRNA组学水平对球囊菌侵染意大利蜜蜂(,意蜂)幼虫和中蜂幼虫过程的宿主与病原相互作用进行了系统探究[17-21],组装并注释了球囊菌的参考转录组[21];基于球囊菌菌丝和孢子混合样品的高质量组学数据鉴定出379个长链非编码RNA和551个环状RNA,并通过分子生物学手段验证了它们的真实表达[22-23]。【本研究切入点】笔者团队前期基于球囊菌菌丝和孢子混合样品的sRNA组学数据鉴定出118个miRNA,并预测和分析了miRNA的靶mRNA及调控网络[24]。然而,球囊菌菌丝和孢子miRNA的数量、结构特征、表达谱差异仍未明确,miRNA与菌丝和孢子生长、发育、生殖和致病性的关系还不清楚。【拟解决的关键问题】明确球囊菌菌丝和孢子miRNA的数量、结构特征和表达谱差异,揭示共有miRNA、特有miRNA和差异表达miRNA(differentially expressed miRNA,DEmiRNA)与菌丝和孢子生长、发育、生殖及致病性的潜在关系。
1 材料与方法
试验于2019年在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。
1.1 供试真菌
球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离和保存[22-24]。
1.2 球囊菌培养及测序样品制备
参照笔者团队前期已建立的方法[19-20,22-23],将实验室保存的球囊菌孢子接种于10块马铃薯葡萄糖琼脂(potato dextrose agar,PDA)固体培养基上,置于33℃生化培养箱中培养,培养7 d的PDA固体培养基上层为蓬松的白色菌丝,覆盖着下层的黑色孢子囊,为避免菌丝和孢子囊之间的交叉污染,首先在超净台中用干净的接种环刮取最上层的白色菌丝,避免接触与孢子囊接触的菌丝,将刮取的菌丝(AaM)集中于一个RNA Free的EP管,经液氮速冻后迅速移至-80℃超低温冰箱保存备用;在超净台中通过无菌操作将覆盖在孢子囊上的薄层菌丝小心刮去,再用另一个干净的接种环将孢子囊刮至一个RNA Free的EP管;继而按照Jensen等[25]的方法进行差速离心,球囊菌孢子密度较大,沉淀在EP管底部,弃去上清;重复上述差速离心3次,弃去上清,保留孢子沉淀;取少量孢子制备悬液,取少量悬液进行显微制片及观察,视野中可见较多的游离孢子,未见菌丝,因而得到的孢子沉淀即为球囊菌的纯净孢子(AaS);将上述EP管投入液氮速冻,然后迅速移至-80℃超低温冰箱保存备用。
1.3 cDNA文库构建及深度测序
(1)用Trizol法分别提取AaM和AaS的总RNA,琼脂糖凝胶电泳切胶选择18—30 nt的片段,分别连接3′接头和5′接头;(2)对连接了两侧接头的small RNA进行反转录和PCR扩增;(3)琼脂糖凝胶电泳回收并纯化约140 bp的条带,完成文库构建。委托广州基迪奥生物科技有限公司对上述样品进行单端测序,测序平台为Illumina MiSeq。测序数据已上传到NCBI SRA数据库,BioProject号:PRJNA560456。
1.4 测序数据质控
按照前期已建立的方法[24,26-27]对下机的原始读段(raw reads)进行质量控制:(1)过滤掉质量值低于20的碱基数超过1个的reads;(2)过滤除掉含有未知碱基(N)的reads;(3)过滤3′或5′接头的reads,并去除长度<18 bp的reads;(4)过滤包含poly A的reads。过滤后得到的clean reads用于后续分析。
1.5 菌丝和孢子miRNA的预测与Venn分析
将严格质控后得到的small RNA有效标签序列(clean tags)分别比对到GenBank数据库(http://www. ncbi.nlm.nih.gov/Web/Genbank/)、Rfam数据库(http://rfam.xfam.org/)、核糖体数据库(http://oberon. fvms.ugent.be:8080/rRNA/lsu/index.html)和球囊菌参考基因组(assembly AAP 1.0,www.ncbi.nlm.nih.gov/ genome/656?genome_assembly_id=274809),以去除可能的rRNA、scRNA、snoRNA、snRNA和tRNA,以及比对上的基因组外显子区域、内含子区域和重复序列区域的clean tags,将未比对上的clean tags与miRBase数据库(www.mirbase.org/)中的miRNA前体序列进行比对,从而鉴定已知miRNA序列。利用Mireap_V 0.2软件将剩余的未比对上的clean tags比对球囊菌参考基因组(assembly AAP 1.0),得到可能的前体序列,根据clean tags在前体序列的分布信息和前体结构能量信息,采用贝叶斯模型打分预测新miRNA。按照TPM=T×106/N(T表示miRNA的tags,N表示总miRNA的tags)算法对AaM和AaS中每个miRNA的表达量进行归一化处理。利用基迪奥在线工具集合(www.omicshare.com)对AaM和AaS的miRNA进行Venn分析,采用默认参数,分别筛选出AaM和AaS的共有miRNA及特有miRNA。
1.6 共有与特有miRNA的靶mRNA的GO和KEGG数据库注释分析
采用RNA hybrid(v2.1.2)+svm_light(v6.01)、Miranda(v3.3a)和TargetScan(Version 7.0)软件[28-31]分别预测共有miRNA和特有miRNA的靶mRNA,将预测结果的交集作为可信度高的靶标集合。利用BLAST工具将靶mRNA比对到GO数据库(http:// geneontology.org/)和KEGG数据库(https://www. kegg.jp/),获得相应的功能和通路注释信息,并统计比对上各功能条目或通路(pathway)的靶mRNA数量。
1.7 DEmiRNA的筛选和靶mRNA的预测及分析
以|log2fold change (FC)|≥1且≤0.05为标准,利用edgeR软件[32]筛选AaM vs AaS的DEmiRNA。前期已利用链特异性建库的RNA-seq技术对球囊菌菌丝和孢子分别进行测序,其中高质量的mRNA组学数据可作为本研究中靶mRNA的数据来源(未发表数据)。分别采用RNA hybrid+svm_light、Miranda和TargetScan软件[28-31]预测DEmiRNA的靶mRNA,将预测结果的交集作为可信度高的靶标集合。利用BLAST工具将DEmiRNA的靶mRNA比对到GO和KEGG数据库,获得相应的功能和通路注释信息,并统计比对上的功能条目和通路的靶mRNA数量。根据上述DEmiRNA与mRNA的靶向结合关系,按照自由能<-40 kcal·mol-1和<0.05的阈值筛选靶向结合关系并构建调控网络,通过Cytoscape软件进行调控网络的可视化。
1.8 DEmiRNA的Stem-loop RT-qPCR验证
随机选取5个DEmiRNA(miR5658-x、miR-10285- y、miR-3245-y、miR4404-x、miR-9-z)进行Stem-loop RT-qPCR验证。参照Chen等[33]和郭睿等[34]的方法,利用DNAMAN软件(Lynnon Biosoft公司,美国)设计上述DEmiRNA的Stem-loop引物、特异性上游引物和通用下游引物。委托上海生工生物工程股份有限公司合成引物(表1)。利用总RNA抽提试剂盒(Promega公司,中国)分别抽提菌丝和孢子的总RNA,然后用RNase-free DNase I去除各自基因组DNA残留。吸取1 μL RNA,用NanoDrop One(Thermo Scientific公司,美国)分别对菌丝、孢子的RNA浓度进行测定。利用Stem-loop引物,按照cDNA第1链合成试剂盒(TaKaRa公司,日本)说明书进行总RNA的反转录,得到的cDNA作为模板进行qPCR。选用球囊菌的(5417)作为内参基因。反应体系为20 μL:SYBR Green Dye 10 μL,特异性上游引物1 μL,通用下游引物1 μL,cDNA模板1 μL,RNA-Free H2O 7 μL。每个反应进行3次技术重复。采用2-ΔΔCt法计算DEmiRNA的相对表达量,然后利用GraphPad Prism 7软件进行Student’s-test检验及绘图。

表1 本研究使用的引物
2 结果
2.1 测序数据的质控与评估
AaM和AaS分别测得12 982 320和12 708 832条raw reads,经严格过滤和质控后分别得到10 800 101和9 888 848条clean tags,占raw reads的比例分别为84.34%和78.83%;此外,比对上参考基因组的clean tags比例分别为80.59%和71.60%(表2)。上述结果表明本研究的sRNA-seq数据质量良好,可满足后续分析。
2.2 球囊菌菌丝和孢子miRNA的预测及比较分析
基于AaM和AaS的高质量数据分别预测出193和275个miRNA。其中,AaM中miRNA的长度介于18—26 nt(图1-A),AaS中miRNA的长度介于18—24 nt(图1-C);AaM和AaS中首位碱基偏向于U的miRNA数量最多,占比分别达到30.77%和45.73%(图1-B、1-D)。
AaM中表达量最高的是miR6478-x、miR10516-x和miR482-x,TPM值分别达到294 585.5787、200 923.7875和99 563.767;AaS中表达量最高的同样为miR6478-x、miR482-x和miR10516-x,TPM值分别为209 814.8692、71 995.2983和71 407.5815。AaM和AaS中表达量最高的前10位miRNA的详细信息如表3和表4所示。
2.3 球囊菌菌丝和孢子共有和特有miRNA的筛选及靶mRNA分析
Venn分析结果显示,AaM和AaS的共有miRNA为76个(19.4%),特有miRNA分别为117个(29.8%)和199个(50.8%)。AaM和AaS的共有miRNA可靶向5 946个mRNA,二者的特有miRNA可分别靶向6 141和6 346个mRNA。GO数据库注释结果显示,上述共有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 027)、细胞(1 027)和细胞器(692),分子功能大类注释靶mRNA数最多的条目是催化活性(1 199)、结合(871)和转运活性(129),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 303)、细胞进程(1 301)和单一组织进程(975);AaM的特有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 095)、细胞(1 095)和细胞器(736),分子功能大类注释靶mRNA数最多的条目是催化活性(1 241)、结合(900)和转运活性(128),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 362)、细胞进程(1 350)和单一组织进程(1 021);AaS的特有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 121)、细胞(1 121)和细胞器(762),分子功能大类注释靶mRNA数最多的条目是催化活性(1 256)、结合(915)和转运活性(126),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 383)、细胞进程(1 379)和单一组织进程(1 039)。括号内的数字表示注释到该条目的靶mRNA数。

表2 sRNA-seq数据概览

A:AaM中miRNA的长度分布Length distribution of miRNAs in AaM;B:AaM中miRNA的首位碱基偏向性 First base bias of miRNAs in AaM;C:AaS中miRNA的长度分布Length distribution of miRNAs in AaS;D:AaS中miRNA的首位碱基偏向性 First base bias of miRNAs in AaS
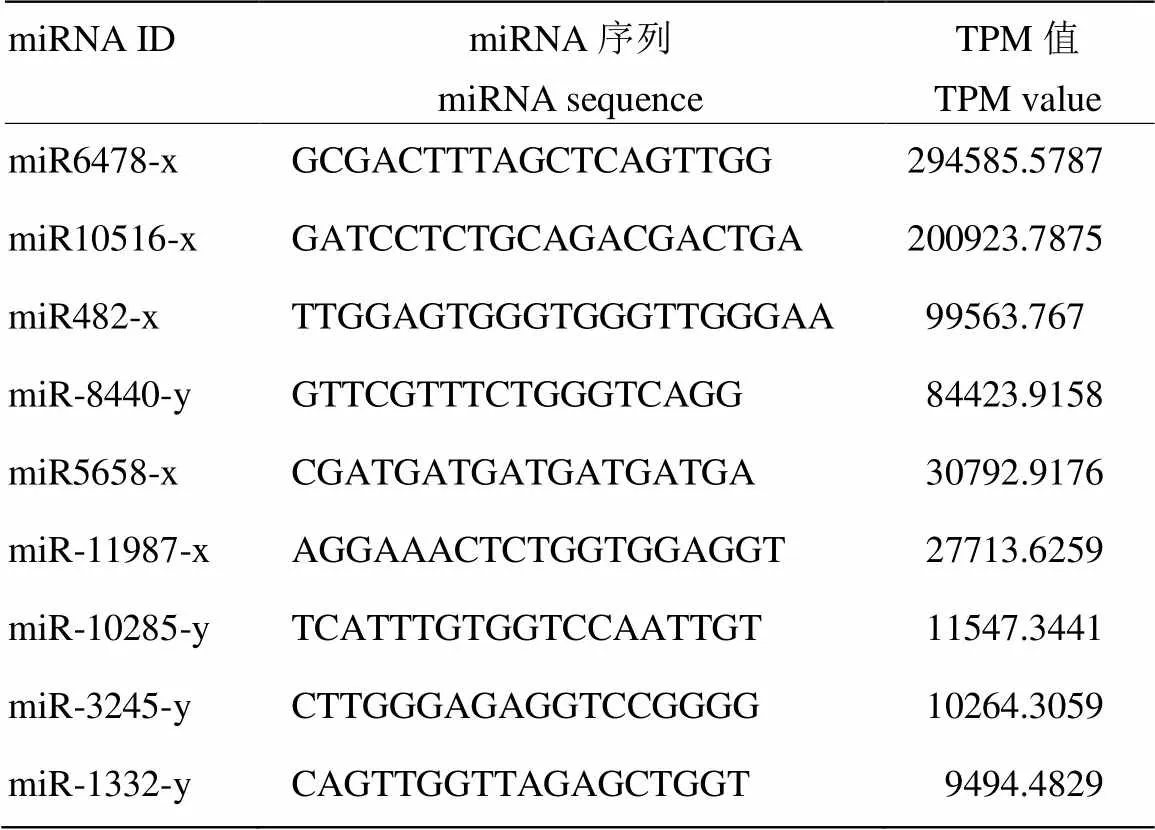
表3 AaM中表达量最高的前10位miRNA

表4 AaS中表达量最高的前10位miRNA
KEGG数据库注释结果显示,上述共有miRNA和二者特有miRNA的靶mRNA均可注释到120条通路,涉及代谢、遗传信息处理、环境信息处理和细胞进程4个大类。其中,共有miRNA的靶mRNA注释数量最多的通路是新陈代谢总览(690)、翻译(264)、碳水化合物代谢(232)、折叠、分类与降解(223)、运输与代谢(186)、传染性疾病(156)、信号转导(138)、能量代谢(133)、细胞生长与死亡(133)和脂质代谢(124);AaM的特有miRNA的靶mRNA注释数量最多的是新陈代谢总览(712)、翻译(283)、碳水化合物代谢(232)、折叠与分类降解(231)、氨基酸代谢(202)、运输与代谢(189)、传染性疾病(164)、信号转导(144)、能量代谢(137)和细胞生长与死亡(134);AaS的特有miRNA的靶mRNA注释数量最多的是新陈代谢总览(725)、翻译(292)、碳水化合物代谢(236)、折叠、分类与降解(236),氨基酸代谢(209)、运输与代谢(194)、传染性疾病(170)、信号转导(148)、细胞生长与死亡(138)和能量代谢(135)。进一步分析发现,AaM的116个特有miRNA靶向的287个mRNA可注释到次级代谢物的生物合成通路;AaS的197个特有miRNA靶向的288个mRNA可注释到次级代谢通路的生物合成通路;44个共有miRNA靶向的14个mRNA可注释到自噬通路;56个共有miRNA靶向的6个mRNA,以及AaM的95个特有miRNA靶向的28个mRNA可注释到MAPK信号通路。括号内的数字表示注释到该通路的靶mRNA数。
2.4 球囊菌菌丝和孢子中miRNA的差异表达分析
从AaM vs AaS中筛选出93个DEmiRNA,包括65个上调miRNA和28个下调miRNA;其中上调幅度最大的是novel-m0040-3p(log2fc=20.65019,=5.98E-17),其次是novel-m0016-3p(log2fc= 20.45754,=3.77E-15)和miR319-y(log2fc=20.23515,=4.75E-13);下调幅度最大的为miR-4028-y(log2fc= -19.6472765,=4.77E-10),其次为miR-4171-x(log2fc=-18.2322391,=0.00049)和miR7787-y(log2fc=-18.1067082,=0.000979)(表5)。上述DEmiRNA能靶向结合6 090个mRNA。
DEmiRNA的靶mRNA可注释到38个功能条目,包括细胞进程(1 268)、代谢进程(1 254)、单一组织进程(981)、定位(332)和生物学调控(290)等15条生物学进程相关条目;催化活性(1 230)、结合(879)、转运活性(125)、结构分子活性(35)及分子功能调节器(18)等11条分子功能相关条目;细胞(1058)、细胞组件(1 058)、细胞器(713)、大分子复合物(390)和细胞膜(332)等12条细胞组分相关条目(图2)。此外,这些靶mRNA可注释到120条通路,注释数量最多的是新陈代谢通路(652)、次级代谢物的生物合成(285)、抗生素的生物合成(212)、微生物在不同环境中的代谢(172)、氨基酸的生物合成(115)、碳代谢(101)、嘌呤代谢(81)、核糖体(78)、剪接体(77)及RNA转运(76)(表6)。括号内的数字代表注释到该条目(通路)的靶mRNA数。

表5 AaM vs AaS比较组中前10位上调和下调miRNA
2.5 DEmiRNA的调控网络构建及分析
根据靶向结合关系构建调控网络,分析结果显示13个DEmiRNA可靶向结合131个mRNA;同一个DEmiRNA可靶向结合多个mRNA,如miR-4968-y可靶向结合多达118个mRNA;同时,部分mRNA可靶向结合1—2个DEmiRNA,如KZZ97168.1分别靶向结合miR-4968-y和miR-6769-y,KZZ91616靶向结合miR5782-y(图3)。
2.6 DEmiRNA的RT-qPCR验证
RT-qPCR结果显示,miR5658-x、miR-10285-y、miR-3245-y、miR4404-x和miR-9-z的表达变化趋势与测序数据相符(图4),证实了本研究中sRNA-seq数据的可靠性。
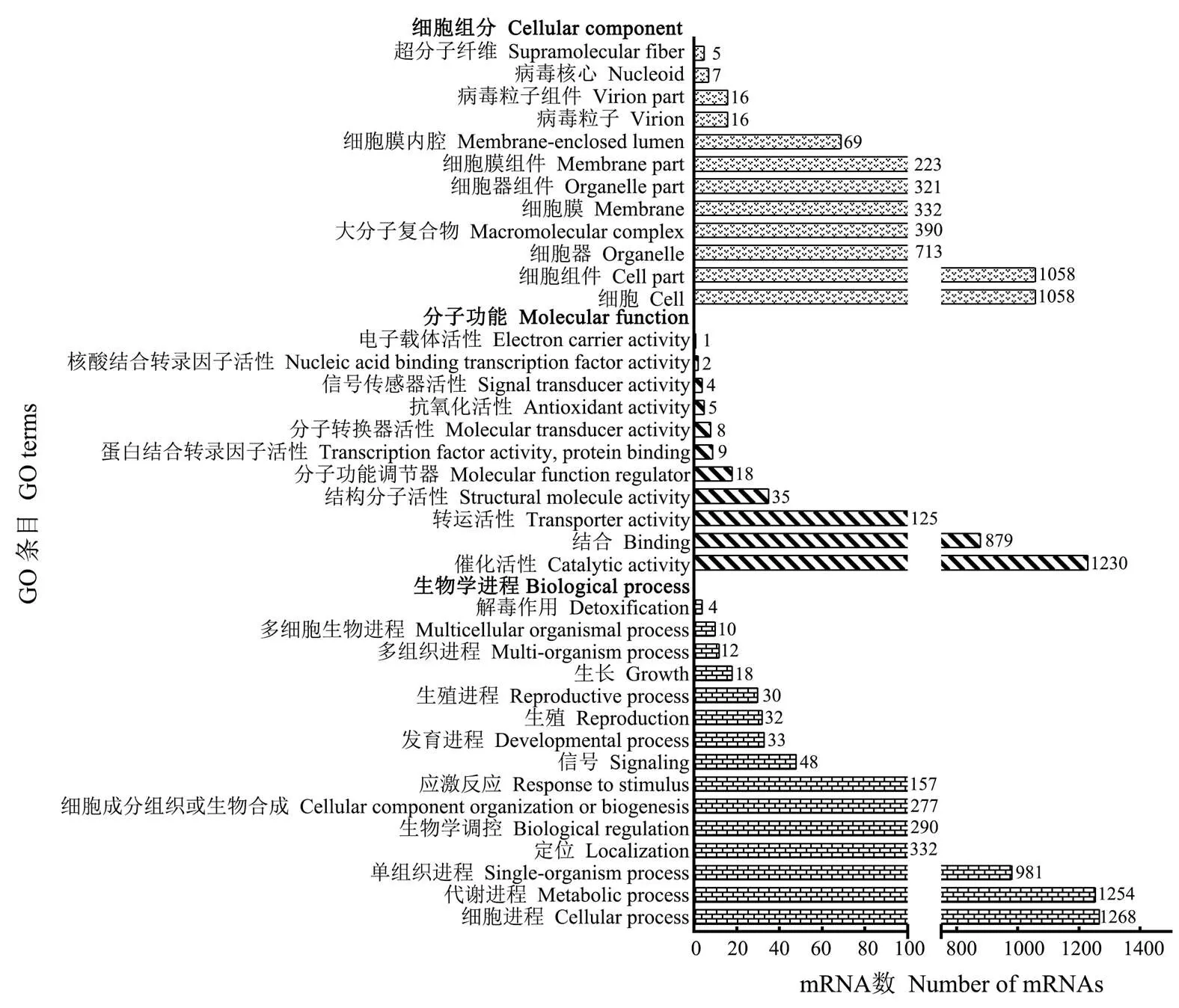
图2 AaM vs AaS比较组中DEmiRNA的靶mRNA的GO数据库注释
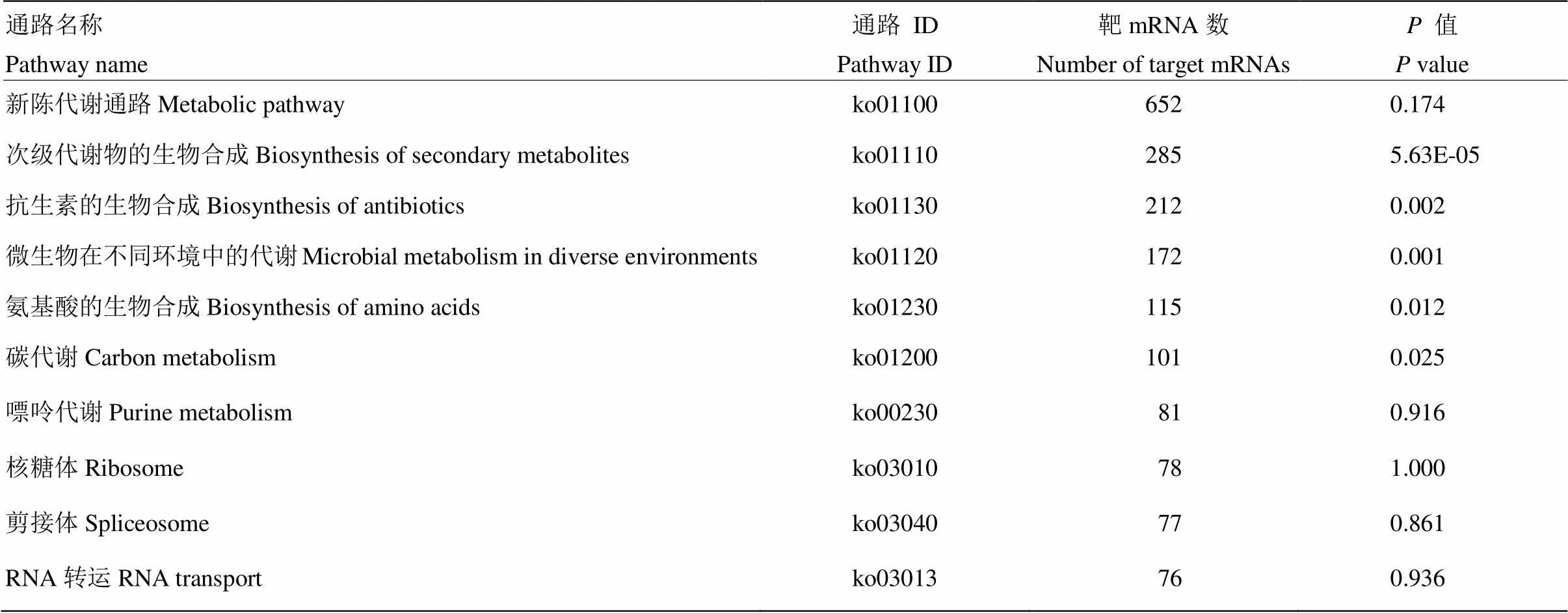
表6 AaM vs AaS比较组中DEmiRNA的靶mRNA注释数前10位通路
3 讨论
MiRNA已被证实能够参与调控真菌的菌丝生长和孢子形成过程[35-38]。例如,Shao等通过比较分析筛选出冬虫夏草()无性生殖阶段和有性生殖阶段的19个DEmiRNA,进而通过过表达和敲除实验证实milR4和milR16参与了菌丝生长过程的调控[36]。前期研究中,为最大限度鉴定miRNA,笔者利用sRNA-seq技术对球囊菌菌丝和孢子的混合样品进行测序,利用miRDeep2软件鉴定到118个novel miRNA,这是关于球囊菌miRNA的首例报道[24]。然而,由于测序得到的混合数据无法区分来源于菌丝的数据和来源于孢子的数据,导致难以进一步对菌丝和孢子中的miRNA进行数量、结构特征、表达谱、靶mRNA及调控网络的比较分析和深入挖掘。因此,本研究首先在实验室条件下获得纯培养的球囊菌,利用sRNA-seq技术对纯净的球囊菌菌丝样品、孢子样品分别进行测序,基于二者的高质量sRNA组学数据分别鉴定到193和275个miRNA,它们的长度分别介于18—26和18—24 nt。分布在18 nt长度的miRNA数量最多,且首位碱基主要偏向U,其结构特征与灰盖鬼伞菌()[37]、新月弯孢()[39]和马尔尼菲青霉()[40]等真菌以及蜜蜂和棉花等动植物[41-42]的miRNA结构高度相似。本研究中,有76个miRNA同时在球囊菌菌丝和孢子中表达,占二者全部miRNA的比例分别为39.38%和27.64%,推测这些共有miRNA在球囊菌的不同生长发育时期均具有一定的调控功能;此外,分别有117和199个miRNA在球囊菌菌丝和孢子中特异性表达,鉴于孢子是球囊菌的休眠态,新陈代谢等生命活动较之菌丝更低,该结果一定程度说明较多的miRNA通过在孢子中特异性表达发挥更强的基因表达抑制(或降解)作用。
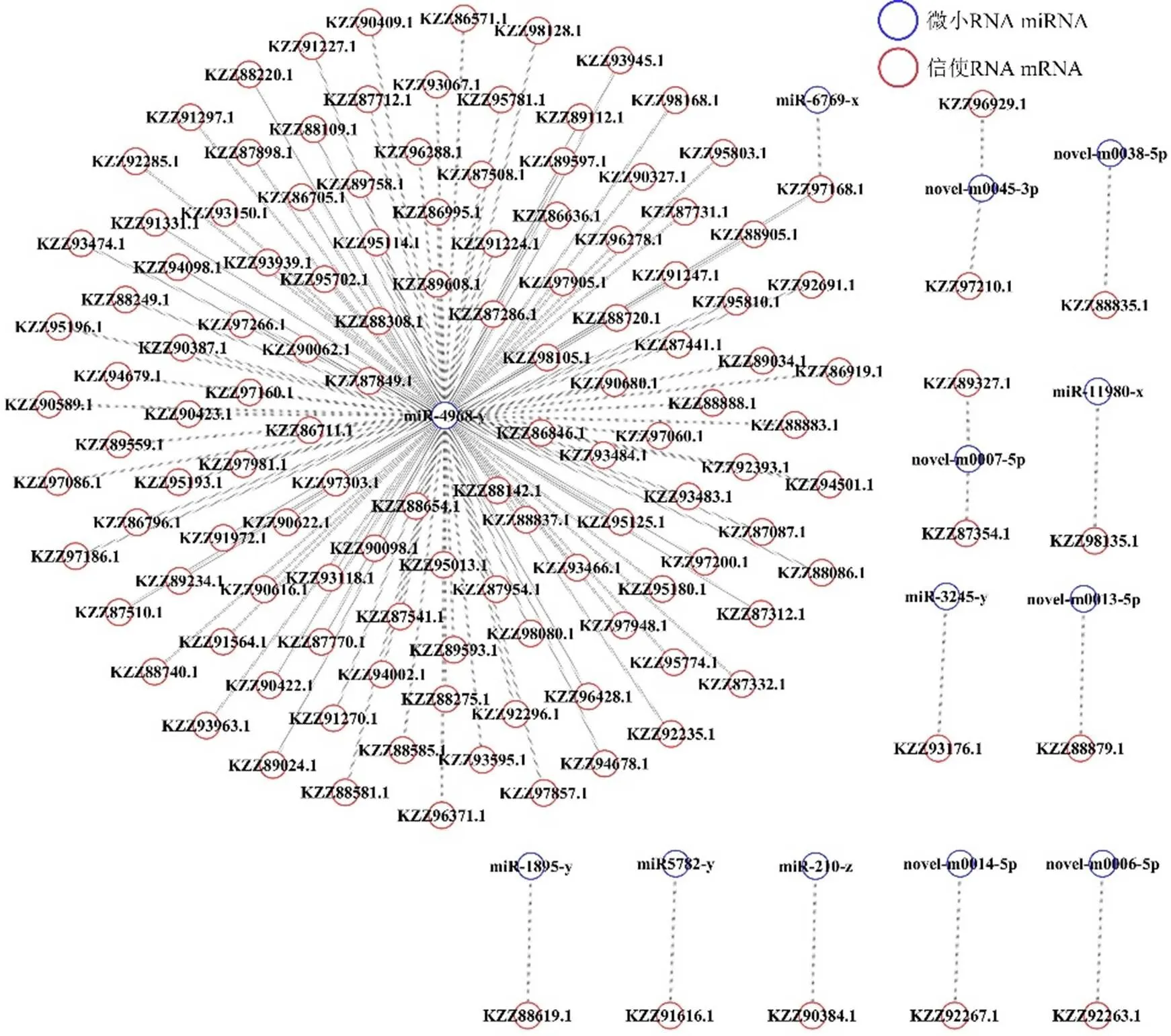
图3 DEmiRNA-mRNA的调控网络

RT-qPCR 组中, *表示P<0.05, **表示P<0.01 In RT-qPCR group, * indicates P<0.05, ** indicates P<0.01
共有miRNA的靶mRNA可注释到代谢进程、生殖、生殖进程、生长等42个功能条目,以及代谢途径、次级代谢物的生物合成、不同环境下微生物的代谢、氨基酸的生物合成、碳代谢及嘌呤代谢和氧化磷酸化等120条通路,表明共有miRNA在球囊菌菌丝和孢子的生长发育、物质和能量代谢、环境适应等方面具有广泛的调控功能。菌丝的特有miRNA可靶向结合6 141个mRNA,这些靶标可注释到42个功能条目和120条通路;孢子的特有miRNA可靶向结合6 346个mRNA,同样可注释到42个功能条目和120条通路。对于真菌孢子中是否存在转录和翻译等生命活动,相关研究报道很少。笔者团队前期通过分子生物学和组学手段证实另一种广泛存在的蜜蜂真菌病原东方蜜蜂微孢子虫()的孢子中存在基因转录[43]。本研究中,在球囊菌孢子中鉴定到199个特有miRNA,上述结果一定程度表明球囊菌孢子中同样存在基因转录以及miRNA介导的基因表达调控现象。此外,共有93个miRNA在菌丝和孢子中差异表达,其中上调miRNA(65)的数量明显多于下调miRNA(28),进一步说明对于球囊菌孢子,除了具有较多的特异性表达miRNA外,其部分miRNA还能通过上调表达量增强对靶基因的抑制(或降解)作用,从而维持较低的新陈代谢水平。推测这对于球囊菌孢子抵抗外界不良环境及长期存活具有重要意义。
真菌在侵染宿主时会分泌一些次级代谢物促进自身增殖并使宿主致死[44-45]。例如,球孢白僵菌()和卵孢白僵菌()合成及分泌的草酸、类草酸晶体和柠檬酸等次级代谢物可协同致死宿主[44]。有研究表明真菌在菌丝状态产生的次级代谢物主要与真菌毒素有关[45]。本研究发现,球囊菌菌丝特有的116个miRNA(miR-11971-y、miR-14-x、miR-12227-y等)的287个靶mRNA(KZZ86592.1、KZZ86645.1和KZZ86652.1等)可注释到次级代谢物的生物合成通路,表明相应的菌丝特有miRNA参与调控次级代谢物的生物合成过程,进而影响球囊菌毒素的合成。真菌病原对昆虫寄主的侵染能力取决于蛋白酶、几丁质酶及脂酶等毒力因子的协同作用,以及菌丝对围食膜、肠壁和表皮的机械破坏力[46]。Cornman等[46]通过同源性比较发现球囊菌包含61个脂酶基因、51个蛋白酶基因和4个几丁质酶基因。本研究分别有16个(miR-14-x、miR11173-y、miR-1002-x等)和9个(miR-12227-y、miR-4171-x、miR-1002-x等)菌丝特有miRNA靶向几丁质酶合成相关的mRNA(KZZ93915.1和KZZ93066.1),暗示这些miRNA参与调控几丁质酶合成,在球囊菌突破宿主几丁质体表过程中发挥重要调控功能。次级代谢物还能影响真菌的孢子萌发[45,47],例如玉米赤霉烯酮和禾谷镰孢()的次级代谢物可诱导禾谷镰孢分生孢子和菌落的产生[47]。本研究发现,197个孢子特有miRNA调控的288个靶mRNA(KZZ86592.1、KZZ86645.1和KZZ86652.1等)注释到了次级代谢物的生物合成,笔者推测孢子特有miRNA可能通过调控次级代谢物合成的相关mRNA影响孢子的萌发。李琼等[48]研究发现,mro-miR-33负调控罗伯茨绿僵菌()的产孢关键基因的表达,敲除mro-miR-33后的表达量明显上调,同时病原的产孢量增加。本研究中,miR-33-x与mro-miR-33高度同源且在孢子中特异性表达,可能参与调控球囊菌的杂交产孢。细胞自噬与丝状真菌的产孢、程序性细胞死亡及致病力紧密相关[49]。在米曲霉()中,、及等自噬基因的功能缺失将影响其产孢过程[50]。此外,有研究表明烟曲霉()也依赖自噬来调控孢子的形成[51]。本研究中,菌丝和孢子的44个共有miRNA的14个靶mRNA(KZZ87046.1、KZZ87260.1和KZZ87338.1等)可注释到自噬通路,推测这些共有miRNA可通过负调控相关靶mRNA调节球囊菌的杂交产孢。
丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)级联反应参与真菌的交配繁殖、致病性、孢子形成以及毒力水平的调控[52-53],其依赖的MAPK激酶可协助病原真菌侵染宿主[54]。前期研究发现,对于侵染意蜂幼虫的球囊菌,有48个差异表达基因富集在MAPK信号通路且表达水平随着球囊菌胁迫时间的延长而显著增强[19];但对于侵染中蜂幼虫的球囊菌,富集在MAPK信号通路的11个差异表达基因均表现为下调趋势[20]。本研究中,菌丝和孢子的56个共有miRNA(let-7-x、miR-10285-y和miR-11980-x等)的靶mRNA,以及菌丝的95个特有miRNA(miR-11971-y、miR-12227-y和miR-14-x等)的28个靶mRNA均可注释到MAPK信号通路,表明上述miRNA与MAPK信号通路具有潜在的调控关系。目前,笔者团队已利用sRNA-seq技术对正常及球囊菌侵染的意蜂幼虫肠道、中蜂幼虫肠道进行测序,下一步将过滤得到侵染两种蜜蜂幼虫的球囊菌的sRNA组学数据,并结合本研究中的球囊菌孢子的sRNA组学数据进行比较分析,更深入地探讨球囊菌对不同抗性蜜蜂幼虫的侵染机制。
本研究中,DEmiRNA与mRNA之间存在较为复杂的调控关系,miR-4968-y可同时被118个mRNA同时靶向结合,表明miR-4968-y处于调控网络的核心位置,可能在球囊菌菌丝和孢子的生长和发育过程发挥关键调控功能,值得进一步深入研究。根癌农杆菌介导(AtMT)的真菌遗传转化体系已成功用于球孢白僵菌、金龟子绿僵菌()和蜡蚧轮枝菌()等昆虫病原真菌的基因功能研究[55-57]。目前,球囊菌的基因功能研究未见报道。利用AtMT技术对本研究筛选出的菌丝和孢子的关键miRNA进行转基因操作,进而探究其在菌丝和孢子生长发育以及病原致病性方面的功能是下一步的工作重点。
4 结论
分别在球囊菌菌丝和孢子中鉴定出193和275个miRNA,二者中特异性表达的miRNA分别为117和199个;菌丝和孢子中的miRNA具有类似的结构特征,但表达谱表现出明显差异;菌丝和孢子可能通过特异性表达和差异表达部分miRNA对其生长、发育和生殖进行调控。
[1] ARONSTEIN K A, MURRAY K D. Chalkbrood disease in honey bees.,2010, 103(Suppl. 1): S20-S29.
[2] MAXFIELD-TAYLOR S A, MUJIC A B, RAO S. First detection of the larval chalkbrood disease pathogen(Ascomycota: Eurotiomycetes: Ascosphaerales) in adult bumble bees., 2015, 10(4): e0124868.
[3] EVISON S E. Chalkbrood: epidemiological perspectives from the host-parasite relationship., 2015, 10: 65-70.
[4] SPILTOIR C F. Life cycle of()., 1955, 42(6): 501-518.
[5] WIJAYAWARDENE N N, HYDE K D, LUMBSCH H T, LIU J K, MAHARACHCHIKUMBURA S S N, EKANAYAKA A H, TIAN Q, PHOOKAMSAK R. Outline of: 2017., 2018, 88: 167-263.
[6] PÖGGELER S. Mating-type genes for classical strain improvements of ascomycetes., 2001, 56(5/6): 589-601.
[7] FLORES J M, SPIVAK M, GUTIERREZ I. Spores ofcontained in wax foundation can infect honeybee brood., 2005, 108(1/2): 141-144.
[8] FLORES J M, GUTIÉRREZ I, ESPEJO R. The role of pollen in chalkbrood disease in: transmission and predisposing conditions., 2005, 97(6): 1171-1176.
[9] BARTEL D P. MicroRNAs: Target recognition and regulatory functions., 2009, 136(2): 215-233.
[10] BURKLEW C E, XIE F U, AshlockJ, ZhangB h. Expression of microRNAs and their targets regulates floral development in tobacco ()., 2014, 14(2): 299-306.
[11] KIDNER C A, MARTIENSSEN R A. Spatially restricted microRNA directs leaf polarity through ARGONAUTE1., 2004, 428(6978): 81-84.
[12] LEE R C, FEINBAUM R L, AMBROS V. Theheterochromatic gene lin-4 encodes small RNAs with antisense complementarity to lin-14., 1993, 75(5): 843-854.
[13] GRIMSON A, SRIVASTAVA M, FAHEY B, WOODCROFT B J, CHIANG H R, KING N, DEGNAN B M, ROKHSAR D S, BARTEL D P. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals., 2008, 455(7217): 1193-1197.
[14] LLAVE C, KASSCHAU K D, RECTOR M A, CARRINGTON J C. Endogenous and silencing-associated small RNAs in plants., 2002, 14(7): 1605-1619.
[15] MOLNÁR A, SCHWACH F, STUDHOLME D J, THUENEMANN E C, BAULCOMBE D C. MiRNAs control gene expression in the single-cell alga., 2007, 447(7148): 1126-1129.
[16] ZHAO T, LI G, MI S, LI S, HANNON G J, WANG X J, QI Y. A complex system of small RNAs in the unicellular green alga., 2007, 21(10): 1190-1203.
[17] CHEN D F, GUO R, XU X J, XIONG C L, LIANG Q, ZHENG Y Z, LUO Q, ZHANG Z N, HUANG Z J, KUMAR D, XI W J, ZOU X, LIU M. Uncovering the immune responses oflarval gut toinfection utilizing transcriptome sequencing., 2017, 621: 40-50.
[18] GUO R, CHEN D F, DIAO Q Y, XIONG C L, ZHENG Y Z, HOU C S. Transcriptomic investigation of immune responses of thelarval gut infected by., 2019, 166: 107210.
[19] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 黄枳腱, 张曌楠, 张璐, 李汶东, 童新宇, 席伟军. 胁迫意大利蜜蜂幼虫肠道的球囊菌的转录组分析. 昆虫学报, 2017, 60(4): 401-411.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, HUANG Z J, ZHANG Z N, ZHANG L, LI W D, TONG X Y, XI W J. Transcriptomic analysis ofstressing larval gut of., 2017, 60(4): 401-411. (in Chinese)
[20] 郭睿, 陈大福, 黄枳腱, 梁勤, 熊翠玲, 徐细建, 郑燕珍, 张曌楠, 解彦玲, 童新宇, 侯志贤, 江亮亮, 刀晨. 球囊菌胁迫中华蜜蜂幼虫肠道过程中病原的转录组学研究. 微生物学报, 2017, 57(12): 1865-1878.
GUO R, CHEN D F, HUANG Z J, LIANG Q, XIONG C L, XU X J, ZHENG Y Z, ZHANG Z N, XIE Y L, TONG X Y, HOU Z X, JIANG L L, DAO C. Transcriptome analysis ofstressing larval gut of., 2017, 57(12): 1865-1878. (in Chinese)
[21] 张曌楠, 熊翠玲, 徐细建, 黄枳腱, 郑燕珍, 骆群, 刘敏, 李汶东, 童新宇, 张琦, 梁勤, 郭睿, 陈大福. 蜜蜂球囊菌的参考转录组组装及SSR分子标记开发. 昆虫学报, 2017, 60(1): 34-44.
ZHANG Z N, XIONG C L, XU X J, HUANG Z J, ZHENG Y Z, LUO Q, LIU M, LI W D, TONG X Y, ZHANG Q, LIANG Q, GUO R, CHEN D F.assembly of a reference transcriptome and development of SSR markers for, 2017, 60(1): 34-44. (in Chinese)
[22] GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, LI W D, KUMAR D, LIANG Q. Identification of long non-coding RNAs in the chalkbrood disease pathogen., 2018, 156: 1-5.
[23] GUO R, CHEN D F, CHEN H Z, FU Z M, XIONG C L, HOU C S, ZHENG Y Z, GUO Y L, WANG H P, DU Y, DIAO Q Y. Systematic investigation of circular RNAs in, a fungal pathogen of honeybee larvae., 2018, 678: 17-22.
[24] 郭睿, 王海朋, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 赵红霞, 陈大福. 蜜蜂球囊菌的microRNA鉴定及其调控网络分析. 微生物学报, 2018, 58(6): 1077-1089.
GUO R, WANG H P, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, ZHAO H X, CHEN D F. Identification ofmicroRNAs and investigation of their regulation networks., 2018, 58(6): 1077-1089. (in Chinese)
[25] JENSEN A B, ARONSTEIN K, FLORES J M, VOJVODIC S, PALACIO M A, SPIVAK M. Standard methods for fungal brood disease research., 2013, 52(1): DOI: 10.3896/IBRA.1.52.1.13.
[26] 郭睿, 杜宇, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 王海朋, 陈华枝, 耿四海, 周丁丁, 石彩云, 赵红霞, 陈大福. 意大利蜜蜂幼虫肠道发育过程中的差异表达microRNA及其调控网络. 中国农业科学, 2018, 51(21): 4197-4209.
GUO R, DU Y, XIONG C L, ZHENG Y Z, FU Z M, XU G J, WANG H P, CHEN H Z, GENG S H, ZHOU D D, SHI C Y, ZHAO H X, CHEN D F. Differentially expressed microRNA and their regulation networks during the developmental process oflarval gut., 2018, 51(21): 4197-4209. (in Chinese)
[27] 杜宇, 范小雪, 蒋海宾, 王杰, 范元婵, 祝智威, 周丁丁, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 陈大福, 郭睿. 微小RNA及其介导的竞争性内源RNA调控网络在意大利蜜蜂工蜂中肠发育过程中的潜在作用. 中国农业科学, 2020, 53(12): 2512-2526.
DU Y, FAN X X, JIANG H B, WANG J, FAN Y C, ZHU Z Z, ZHOU D D, WAN J Q, LU J X, XIONG C L, ZHENG Y Z, CHEN D F, GUO R. The potential role of microRNAs and microRNA-mediated competitive endogenous networks during the developmental process ofa worker’s midgut., 2020, 53(12): 2512-2526. (in Chinese)
[28] RITCHIE W. microRNA target prediction., 2017, 1513: 193-200.
[29] REHMSMEIER M, STEFFEN P, HOCHSMANN M, GIEGERICH R. Fast and effective prediction of microRNA/target duplexes., 2004, 10(10): 1507-1517.
[30] KRÜGER J, REHMSMEIER M. RNAhybrid: microRNA target prediction easy, fast and flexible., 2006, 34: W451-W454.
[31] ALLEN E, XIE Z x, GUSTAFSON A M, CARRINGTON J C. microRNA-directed phasing during-acting siRNA biogenesis in plants., 2005, 121(2): 207-221.
[32] ROBINSON M D, MCCARTHY D J, SMYTH G K. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data., 2010, 26(1): 139-140.
[33] CHEN C F, RIDZON D A, BROOMER A J, ZHOU Z H, LEE D H, NGUYEN J T, BARBISIN M, XU N L, MAHUVAKAR V R, ANDERSEN M R, LAO K Q, LIVAK K J, GUEGLER K J. Real-time quantification of microRNAs by stem-loop RT-PCR., 2005, 33(20): e179.
[34] 郭睿, 杜宇, 周倪红, 刘思亚, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 王海朋, 耿四海, 周丁丁, 陈大福. 意大利蜜蜂幼虫肠道在球囊菌胁迫后期的差异表达微小RNA及其靶基因分析. 昆虫学报, 2019, 62(1): 49-60.
GUO R, DU Y, ZHOU N H, LIU S Y, XIONG C L, ZHENG Y Z, FU Z M, XU G J, WANG H P, GENG S H, ZHOU D D, CHEN D F. Comprehensive analysis of differentially expressed microRNAs and their target genes in the larval gut ofduring the late stage ofstress., 2019, 62(1): 49-60. (in Chinese)
[35] ZHOU Q, WANG Z X, ZHANG J, MENG H M, HUANG B. Genome-wide identification and profiling of microRNA-like RNAs fromduring development., 2012, 116(11): 1156-1162.
[36] SHAO Y, TANG J, CHEN S L, WU Y H, WANG K, MA B, ZHOU Q M, CHEN A H, WANG Y L. milR4 and milR16 mediated fruiting body development in the medicinal fungus., 2019, 10: 83.
[37] LAU A Y T, CHENG X, CHENG C K, NONG W, CHEUNG M K, CHAN R H, HUI J H L, KWAN H S. Discovery of microRNA-like RNAs during early fruiting body development in the model mushroom., 2018, 13(9): e0198234.
[38] ZENG W P, WANG J, WANG Y, LING J, FU Y P, XIE J T, JIANG D H, CHEN T, LIU H Q, CHENG J S. Dicer-like proteins regulate sexual development via the biogenesis of perithecium-specific microRNAs in a plant pathogenic fungus., 2018, 9: 818.
[39] LIU T, HU J, ZUO Y, JIN Y, HOU J. Identification of microRNA-like RNAs fromassociated with maize leaf spot by bioinformation analysis and deep sequencing., 2016, 291(2): 587-596.
[40] LAU S K, CHOW W N, WONG A Y, YEUNG J M, BAO J, ZHANG N, LOK S, WOO P C, YUEN K Y. Identification of microRNA-Like RNAs in mycelial and yeast phases of the thermal dimorphic fungus., 2013, 7(8): e2398.
[41] LI J, HULL J J, LIANG S, WANG Q, CHEN L, ZHANG Q, WANG M, MANSOOR S, ZHANG X, JIN S. Genome-wide analysis of cotton miRNAs during whitefly infestation offers new insights into plant-herbivore interaction.,2019, 20(21): e5357.
[42] 熊翠玲, 杜宇, 陈大福, 郑燕珍, 付中民, 王海朋, 耿四海, 陈华枝, 周丁丁, 吴素珍, 石彩云, 郭睿. 意大利蜜蜂幼虫肠道的miRNAs的生物信息学预测及分析. 应用昆虫学报, 2018, 55(6): 1023-1033.
XIONG C L, DU Y, CHEN D F, ZHENG Y Z, FU Z M, WANG H P, GENG S H, CHEN H Z, ZHOU D D, WU S Z, SHI C Y, GUO R. Bioinformatic prediction and analysis of miRNAs inthelarval gut., 2018, 55(6): 1023-1033. (in Chinese)
[43] GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, LIANG Q, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, KUMAR D. First identification of long non-coding RNAs in fungal parasite.,2018, 49(5): 660-670.
[44] 黄居敏, 张普照, 王芳, 李旸, 冯少俊, 杨明. 白僵菌的代谢产物及药理活性研究进展. 中国生化药物杂志, 2014, 34(9): 167-173.
HUANG J M, ZHANG P Z, WANG F, LI y, FENG S J, YANG MAdvanced studies on metabolites and pharmacological of.,2014, 34(9): 167-173. (in Chinese)
[45] CALVO A M, WILSON R A, BOK J W, KELLER N PRelationship between secondary metabolism and fungal development,2002, 66(3): 447-459.
[46] CORNMAN R S, BENNETT A K, MURRAY K D, EVANS J D, ELSIK C G, ARONSTEIN K. Transcriptome analysis of the honey bee fungal pathogen,: implications for host pathogenesis., 2012, 13: 285.
[47] WOLF J C, MIROCHA C J. Regulation of sexual reproduction in(“Graminearum”) by F-2 (Zearalenone)., 1973, 19(6): 725-734.
[48] 李琼, 崔春来, 宋红生, 王四宝. mro-miR-33在绿僵菌产孢中的作用. 菌物学报, 2017, 36(6): 671-678.
LI Q, CUI C L, SONG H S, WANG S B. The effects of mro-miR-33 on the conidial production in.,2017, 36(6): 671-678. (in Chinese)
[49] 闫思源, 姜学军. 细胞自噬及真菌中自噬研究概述. 菌物学报, 2015, 34(5): 871-879.
YAN S Y, JIANG X J. Overview of autophagy and related research in fungi., 2015, 34(5): 871-879. (in Chinese)
[50] KIKUMA T, KITAMOTO K. Analysis of autophagy inby disruption of Aoatg13, Aoatg4, and Aoatg15 genes., 2011, 316(1): 61-69.
[51] RICHIE D L, FULLER K K, FORTWENDEL J, MILEY M D, MCCARTHY J W, FELDMESSER M, RHODES J C, ASKEW D S. Unexpected link between metal ion deficiency and autophagy in., 2007, 6(12):2437-2447.
[52] ZHAO X, MEHRABI R, XU J R. Mitogen-activated protein kinase pathways and fungal pathogenesis., 2007, 6(10): 1701-1714.
[53] IGBARIA A, LEV S, ROSE M S, LEE B N, HADAR R, DEGANI O, HORWITZ B A. Distinct and combined roles of the MAP kinases ofin virulence and stress responses., 2008, 21(6): 769-780.
[54] CHEN X X, XU C, QIAN Y, LIU R, ZHANG Q Q, ZENG G H, ZHANG X, ZHAO H, FANG W G. MAPK cascade-mediated regulation of pathogenicity, conidiation and tolerance to abiotic stresses in the entomopathogenic fungus., 2016, 18(3): 1048-1062.
[55] LECLERQUE A, WAN H, ABSCHÜTZ A, CHEN S, MITINA G V, ZIMMERMANN G, SCHAIRER H.-mediated insertional mutagenesis (AIM) of the entomopathogenic fungus., 2004, 45(2): 111-119.
[56] FANG W, PEI Y, BIDOCHKA M J. Transformation ofmediated by., 2006, 52(7): 623-626.
[57] ZHANG Y J, ZHAO J J, XIE M, PENG D L.-mediated transformation in the entomopathogenic fungusand development of benzimidazole fungicide resistant strains.,2014, 105: 168-173.
Comparative Analysis of microRNAs and corresponding target mRNAs inmycelium and spore
CHEN HuaZhi, ZHU ZhiWei, JIANG HaiBin, WANG Jie, FAN YuanChan, FAN XiaoXue, WAN JieQi, LU JiaXuan, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui
(College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002)
【】exclusively infects honeybee larvae, resulting in chalkbrood disease. The objective of this study is to clarify the differences of number, structure and expression pattern of miRNAs betweenmycelium and spore based on deep sequencing and comparative analysis of purified mycelia (AaM) and spores (AaS) using small RNA-seq (sRNA-seq) and bioinformatics, and reveal the potential relationship between common miRNAs, specific miRNAs, differentially expressed miRNAs (DEmiRNAs) and their target mRNAs and the growth and development of mycelium and spore as well as pathogenesis of【】The pure culture ofwas gained under lab condition. AaM and AaS were respectively sequenced using sRNA-seq technology. Clean tags were obtained after filtration and quality control of raw reads. Common miRNAs and specific miRNAs in AaM and AaS were screened out using Venn analysis. DEmiRNAs in the AaM vs AaS comparison group were filtered out following the criteria of≤0.05 and |log2fold change|≥1. Target mRNAs of common miRNAs, specific miRNAs and DEmiRNAs were predicted using related bioinformatic software. Target mRNAs mentioned above were respectively annotated to GO database and KEGG database. The regulatory network of DEmiRNAs and target mRNAs was constructed on basis of target binding relationship, followed by visualization with Cytoscape. RT-qPCR was conducted to verify the reliability of the sequencing data.【】In total, 12 982 320 and 12 708 832 raw reads were produced from AaM and AaS, and after strict quality control, 10 800 101 and 9 888 848 clean tags were gained, respectively. The length of specific miRNAs in AaM was distributed among 18-26 nt, while that in AaS was distributed among 18-24 nt. Additionally, most of the miRNAs were distributed in 18 nt. MiRNAs with the first base U in both AaM and AaS were the most abundant. miRNAs with the highest expression levels in both AaM and AaS were miR6478-x, miR10516-x and miR482-x. These common miRNAs could target 5 946 mRNAs, while specific miRNAs in AaM and AaS could bind to 6 141 and 6 346 mRNAs, respectively. Targets of common miRNAs were annotated to 42 functional terms such as metabolism process, cellular process and catalytic activity, and 120 pathways including translation, carbohydrate metabolism and energy metabolism. In addition, a total of 93 DEmiRNAs were identified in AaM vs AaS comparison group, targeting 6 090 mRNAs annotated to 38 functional terms and 120 pathways. Moreover, complicated regulatory networks were formed between DEmiRNAs and target mRNAs, with miR-4968-y located in the center and linked to as many as 118 mRNAs. RT-qPCR result demonstrated the expression trend of five DEmiRNAs was consistent with that in the sequencing result, confirming the reliability of our sequencing data.【】The structures of miRNAs inmycelium and spore were similar, whereas their expression patterns were obviously different; mycelium and spore may specifically and differentially express part of miRNAs to regulate their growth, development and reproduction.
; mycelium; spore; miRNA; mRNA; target binding

10.3864/j.issn.0578-1752.2020.17.017
2019-12-25;
2020-02-04
国家自然科学基金(31702190)、国家现代农业产业技术体系建设专项(CARS-44-KXJ7)、福建省自然科学基金(2018J05042)、福建省教育厅中青年教师教育科研项目(JAT170158)、福建农林大学杰出青年科研人才计划(xjq201814)、福建农林大学科技创新专项(CXZX2017342, CXZX2017343)、福建农林大学优秀硕士学位论文资助基金(陈华枝)
陈华枝,E-mail:CHZ0720@outlook.com。祝智威,E-mail:zzw15235470398@163.com。陈华枝和祝智威为同等贡献作者。通信作者郭睿,E-mail:ruiguo@fafu.edu.cn
(责任编辑 岳梅)
猜你喜欢
陕西农业科学(2022年2期)2022-03-28 02:41:54
浙江农业学报(2021年5期)2021-05-31 07:29:08
中国民间疗法(2021年1期)2021-04-20 02:31:16
上海农业学报(2017年4期)2017-04-10 12:40:22
广东饲料(2016年5期)2016-12-01 03:43:21
渔业致富指南(2016年12期)2016-11-11 07:07:18
实用临床医学(2016年8期)2016-06-07 01:28:24
山东医药(2015年14期)2016-01-12 00:39:55
科学大众(中学)(2015年9期)2015-10-12 09:37:53
少儿科学周刊·少年版(2015年3期)2015-07-07 19:30:33
