
基于高通量测序分析白云岩喀斯特土壤微生物多样性
2020-09-11 04:57李欲轲何馨竹汤晓辛
贵州师范大学学报(自然科学版) 2020年5期
唐 婧,李欲轲,何馨竹,汤晓辛
(贵州师范大学 贵州省植物生理与发育重点实验室/生命科学学院,贵州 贵阳 550001)
0 引言
中国是一个岩溶大国,喀斯特区域达346.3万km2[1]。贵州因地处西南喀斯特的核心区域,故喀斯特地貌最为典型。由于喀斯特地区生态系统脆弱、土壤层浅薄,易出现生态系统退化[2]及“石漠化现象”(石漠化作为我国土地荒漠化的主要类型之一,并以每年2 500 km2的速度不断扩展[3]),不仅制约了西南地区经济的可持续发展,也增加了当地生态系统恢复的难度。
土壤微生物作为土壤生态系统的重要组成部分,不仅在生物化学循环、有机物质分解等方面起着至关重要的作用,与土壤性质、地上植被之间也具有十分密切的关系[4-6]。如:喀斯特生态系统的改变会造成土壤肥力降低[7]及土壤微生物生物量降低[8-9];土壤微生物生物量可作为指示生态系统退化的指示器[10];土壤微生物多样性指标可视为最具潜力的敏感性生物指标之一[11]。可见,土壤微生物群落与生态环境之间具有十分密切的关系。以贵州施秉为典型代表的白云岩喀斯特[12]是世界热带-亚热带喀斯特发育类型的杰出代表,其地貌演化环境独特、地质演化过程复杂、地貌组合形态丰富及生态保护完好,具有极高的研究价值。然而,对白云岩喀斯特土壤微生物的种类和多样性研究目前却少见报道。
鉴于此,本研究基于16S RNA V4测序分析,揭示白云岩喀斯特微生物群落结构特点。同时,利用PICRUSt算法预测微生物群落功能基因组,初步解析白云岩喀斯特土壤微生物功能,以期为白云岩喀斯特土壤微生物群落结构监控提供参考。
1 材料与方法
1.1 样品采集
实验的11个土壤样品均采自施秉白云岩喀斯特地区,每个土壤样点直线相隔距离大于200 m,每个样点由1 m2内5点取样混合收集土壤样品,4 ℃冰箱运输回实验室,后置于-80 ℃冰箱中冻存。
1.2 微生物总DNA的提取
称取土壤样品0.5 g,用PowerSoil DNA Isolation Kit 试剂盒提取宏基因组DNA,使用方法参见说明,提取后的DNA于-80 ℃冰箱中冻存。
1.3 土壤微生物群落结构分析
以提取的宏基因组DNA为模板,分别扩增16S rRNA V4区及ITS1区,利用Illumina平台高通量测序。测序下机数据在QIIME[13]软件平台进行质量控制分析,过滤除去低质量及有嵌合体序列,然后进行合并。利用Ucluster算法[14]进行同源聚类,相似度≥97%的序列划为OTUs。然后基于Rdp算法[15]根据Greengene数据库[16]进行分类注释。利用R的Vegan、Ggplots等软件包分析土壤微生物群落的α-多样性指数(Shannon指数、Simpson指数、Chao1指数及ACE)及稀释曲线。统计分析土壤微生物群落各分类水平的相对丰度,绘制微生物群落分类树。根据皮尔逊相关系数分析土壤性质与土壤微生物群落的相互关系。
1.4 土壤微生物基因功能预测
利用Picrust[17]算法,基于KEGG数据库预测白云岩喀斯特土壤微生物功能基因组,基于皮尔逊相关系数分析土壤性质与土壤微生物功能之间的关系。
2 结果与分析
2.1 样品测序分析
本研究针对施秉白云岩喀斯特地区采集的11个土壤样品提取宏基因组DNA;基于Illumina平台,对16S rRNA V4区及ITS1区进行高通量测序;并对测序下机数据进行严格的质控处理,以97%的序列相似度作为分类阈值,对OTUs聚类。共获取901 320条16S rRNA V4 序列及684 488条ITS1序列,测序数据平均有效率分别为95.65%(±0.94)和90.11%(±2.58)。测序数据的有效率都高于90%,测序数据较好且可靠,细菌16S rRNA V4区测序数据效率较高于真菌ITS1区。OTUs聚类表明,有效序列聚类后分别获得6 387个和3 664个OTUs。绘制稀释曲线(见图1),由图1可见:在该深度测序下,测序数据量足够大,可以基本覆盖到样品中的绝大多数微生物物种,能够很好的反映出样品中微生物的群落结构和多样性。
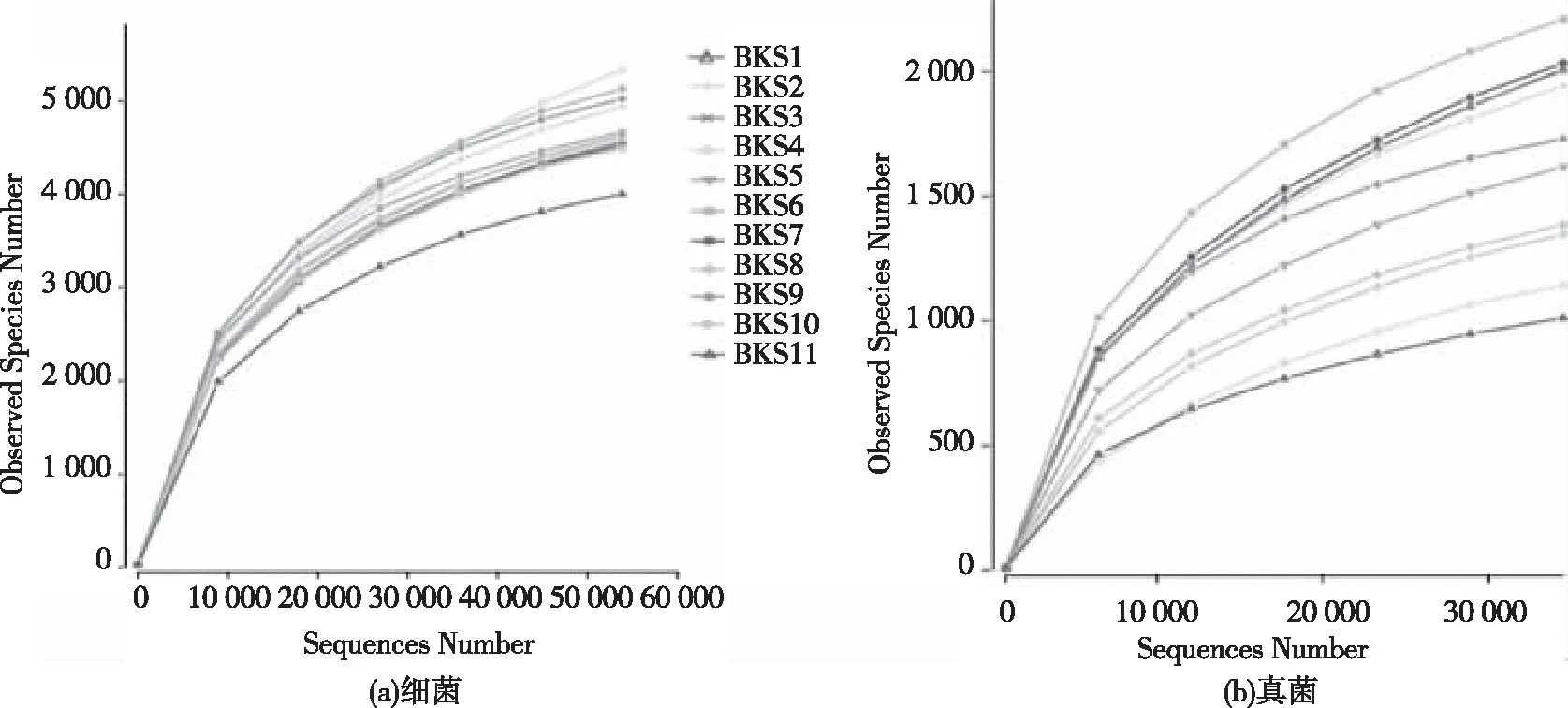
图1 稀释曲线Fig.1 Rarefaction curves of observed species number
2.2 土壤微生物群落结构
2.2.1 细菌物种注释
在各分类水平上对样品进行物种注释分析,构建细菌分类树,以直观展现出细菌群落的结构组成。如图2所示,在门的水平上,相对丰度高于1%的优势菌门主要包括:变形菌门、酸杆菌门、放线菌门、厚壁菌门(Firmicutes)、浮霉菌门(Planctomycetes)、芽单胞菌门(Gemmatimonadetes)、拟杆菌门(Bacteroidetes)、消化螺旋菌门(Nitrospirae)、绿弯菌门(Chloroflexi)、疣微菌门(Verrucomicrobia)以及Latescibacteria共11个菌门。其中主要的优势菌门是:变形菌门(33.5%±2.8)、酸杆菌门(18.4%±1.5)、放线菌门(16.6%±1.9),其物种组成和多样性也最高。细菌基于科的水平上进行比较分析发现,丰度高于1%的共有11个:分别是鞘脂单胞菌科(Sphingomonadaceae)1.5%、黄单胞菌科(Xanthomonadaceae)1.2%、丛毛单胞菌科(Comamonadaceae)1.6%、亚硝化单胞菌科(Nitrosomonadaceae)1.7%、RB41 1.6%、Gaiellaceae 1.5%、乳杆菌科(Lactobacillaceae)1.5%、毛螺旋菌科(Lachnospiraceae)1.1%、浮霉状菌科(Planctomycetaceae)2.5%、芽单胞菌科(Gemmatimonadaceae)3.0%和0319-6A21 2.5%,它们来自于8个菌门。在白云岩喀斯特土壤细菌群落中,含量丰度最高的是鞘氨醇单胞菌属(Sphingomonas, 1.5%)、Gaiella (1.5%) 和乳杆菌属(Lactobacillus, 1.5%)。
2.2.2 真菌群落结构分析
由图3可知:基于门的分类水平分析,白云岩喀斯特土壤真菌群落主要包含4个菌门:子囊菌门、壶菌门(Chytridiomycota)、担子菌门和接合菌门。其丰度依次为53.2%±4.8、2.4%±0.7、18.2%±3.1、25.1%±3.5,其中以子囊菌门丰度最高,其物种组成也最为丰富,多样性也最高。在所有的真菌群落中,丰度前5的科为:被孢霉科(Mortierellaceae)10.1%、(Archaeorhizomycetaceae)5.8%、丛赤壳科(Nectriaceae)4.7%、伞菌科(Agaricaceae)3.9%和(Trichosporonaceae)3.3%。白云岩喀斯特土壤真菌主要包括尾柄孢壳菌属(Cercophora)1.0%、镰刀菌属(Fusarium)1.1%、枝孢菌属(Cladosporium)1.2%、花顶孢酶属(Idriella)1.3%、葡萄孢属(Botrytis)1.3%和(Lycogalopsis) 3.9%的菌群。
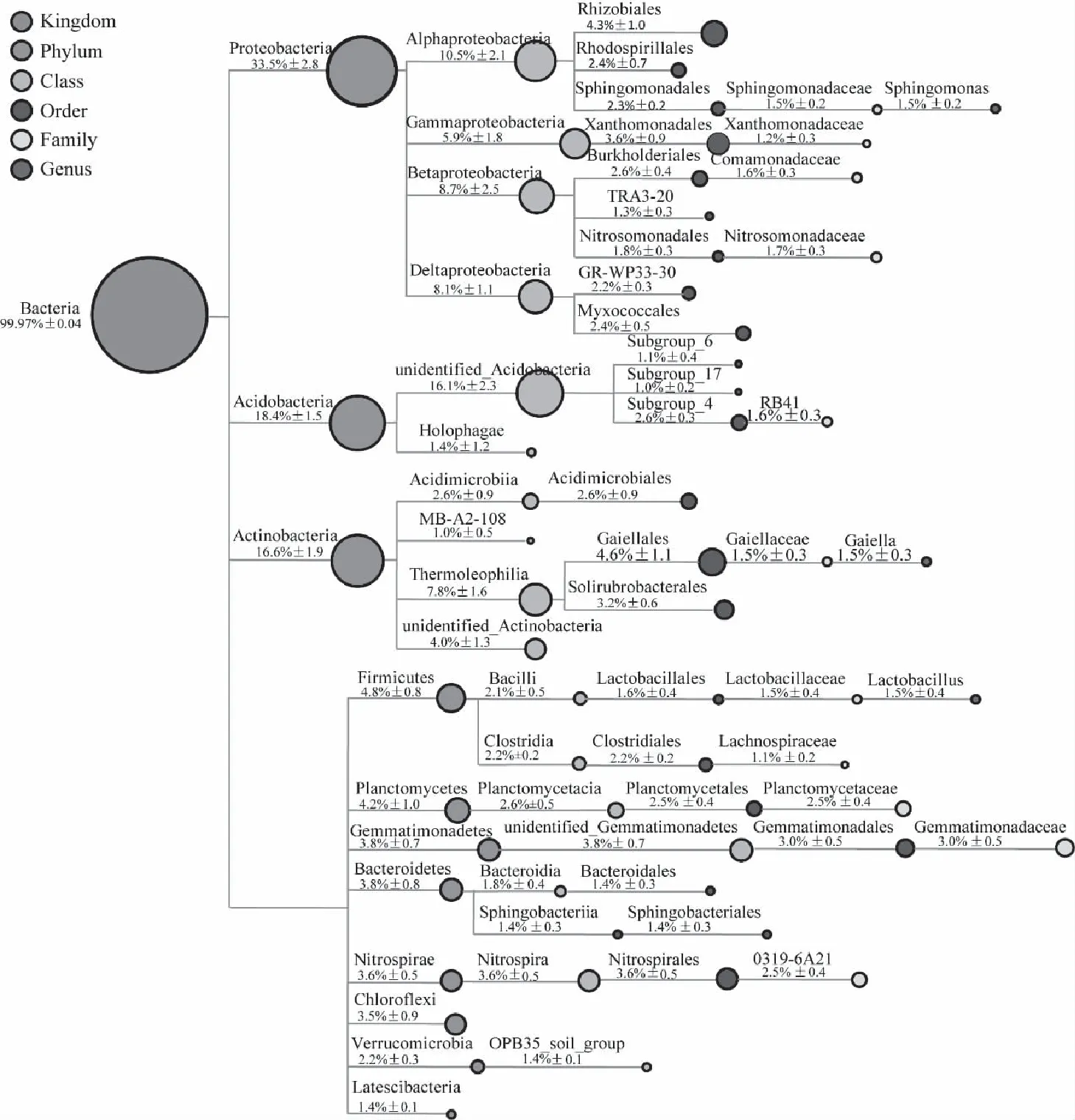
注: 相对丰度高于1%的物种。图2 细菌分类树Fig.2 Taxtree of bacteria
2.2.3 Alpha多样性分析
Alpha多样性指数作为样品中物种组成研究的重要指标,也是一种最基本的多样性研究指数。由表1可见:表明白云岩喀斯特土壤微生物中细菌的α-多样性指数均高于真菌的,说明喀斯特土壤中的细菌群落多样性高于真菌。

表1 Alpha多样性指数Tab.1 The diversity index of Alpha
2.3 土壤微生物群落与土壤性质的关系
利用Mantel test分析白云岩喀斯特土壤微生物群落与土壤性质之间的相关性。结果表明(见图4):在白云岩喀斯特土壤中,土壤微生物群落与土壤性质之间显著相关(P=0.034,P=0.041)。进一步利用皮尔逊相关系数分析不同门类微生物群落与土壤性质之间的相互关系,结果显示(见图4):13个细菌门类都与土壤交换镁(Emg2+)存在相关性。其中,有10个门类与其表现出负相关关系,特别是Hydrogenedentes、螺旋体门(Spirochaetes)、黏胶球形菌门(Lentisphaerae)与Emg2+之间存在显著的负相关关系。而放线菌门、消化螺旋菌门则与Emg2+之间呈显著的正相关关系。真菌门类与Emg2+之间并未表现出明显的相关性。Hydrogenedentes不仅与Emg2+之间存在显著的负相关关系,还与pH显著负相关。此外,绿菌门(Chlorobi)和柔膜菌门(Tenericutes)也和pH呈显著负相关关系。嗜热丝菌门(Caldiserica)、担子菌门与其则是显著正相关。在与土壤总钾(TK)存在相关关系的微生物群落中,有5个菌群门类与其存在显著的相关性。其中,与TK存在显著正相关的菌群是疣微菌门和酸杆菌门;而TM6、Gracilibacteria和变形菌门与TK之间则是显著的负相关关系。Hydrogenedentes、绿菌门和柔膜菌门与pH呈显著负相关关系,却与土壤交换钙(ECa2+)之间显著正相关。此外,绿菌门与土壤有机碳(SOC)、土壤总磷(STP),接合菌门与ECa2+、STP均为显著正相关关系。而嗜热丝菌门与ECa2+,担子菌门与ECa2+、STP之间则表现为显著的负相关。此外,由图4还可知,细菌门类均与Emg2+存在相关性,且大多数门类与其呈现负相关关系,而ECa2+却与之相反,大多微生物群落与其呈正相关关系。
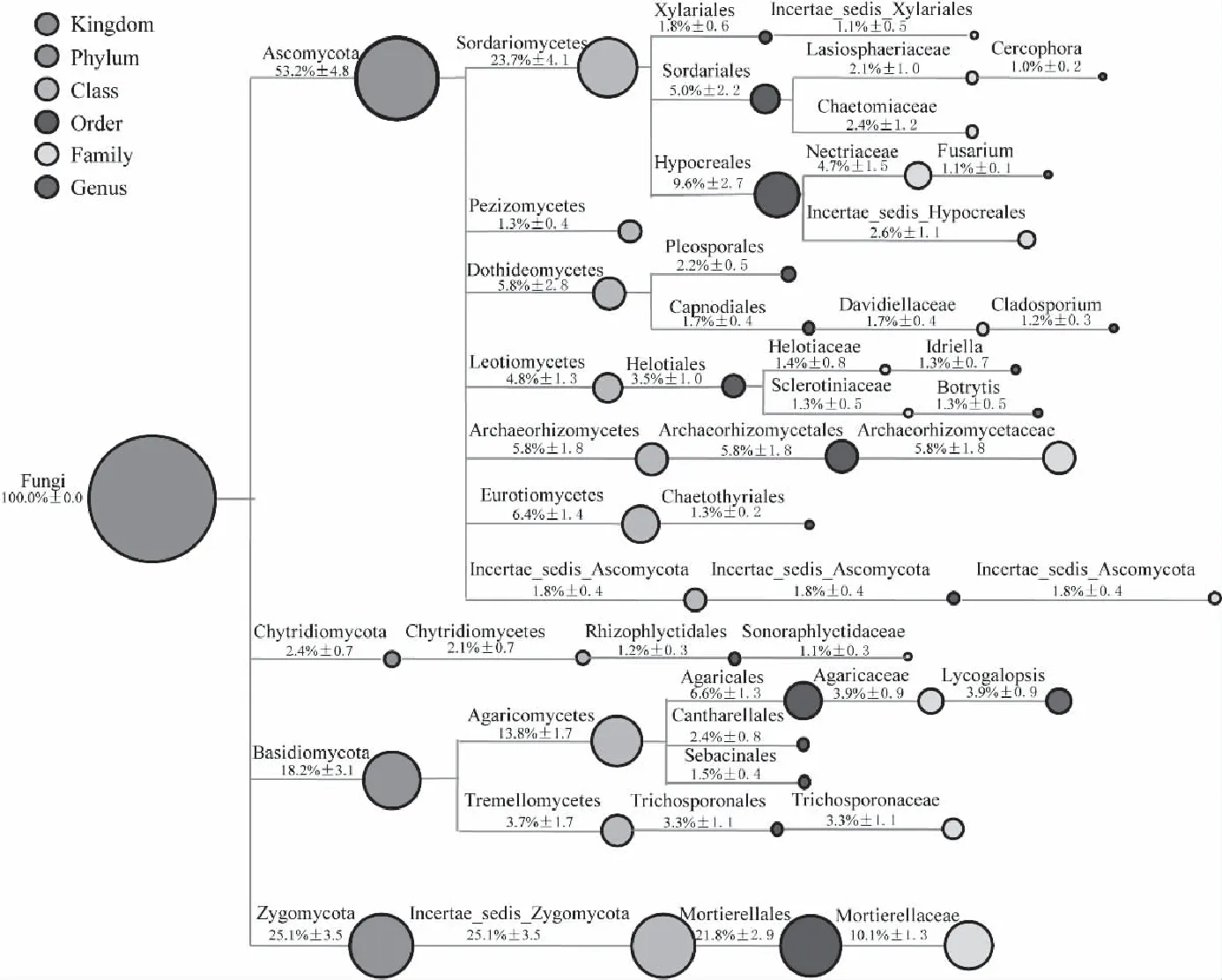
注: 相对丰度高于1%的物种。图3 真菌分类树Fig.3 Taxtree of fungi
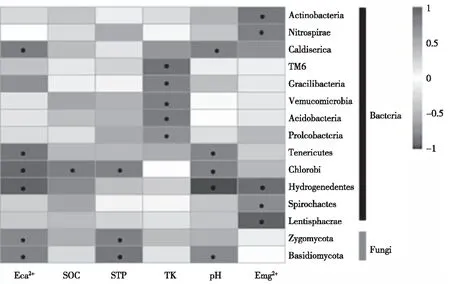
注:*代表显著性相关,下同。图4 土壤性质与微生物群落之间的相关性Fig.4 The relativity between soil property and microbial community
2.4 土壤性质与细菌代谢功能之间的相互作用
2.4.1 PICRUSt基因预测
PICRUSt使用原理在于根据微生物群落的分类信息及微生物群落的丰度数据,利用数据库分析,预测生物群落的功能基因组。本研究通过Picrust算法,基于KEGG数据库,对白云岩喀斯特土壤微生物基因功能进行了预测。结果共预测出6 208个同源功能基因(KEGG orthology),富集于311条代谢通路(pathway)(见图5)。环境信息处理通路中的膜转运信号通路上富集的基因数量最高。有关代谢的基因在种类和数量上,都占据着预测功能基因中的主要地位;其中,有关氨基酸代谢和碳水化合物代谢的通路在富集基因数量上也占绝对显著优势,推测认为在喀斯特土壤中大量细菌进行着活跃的生长代谢活动。
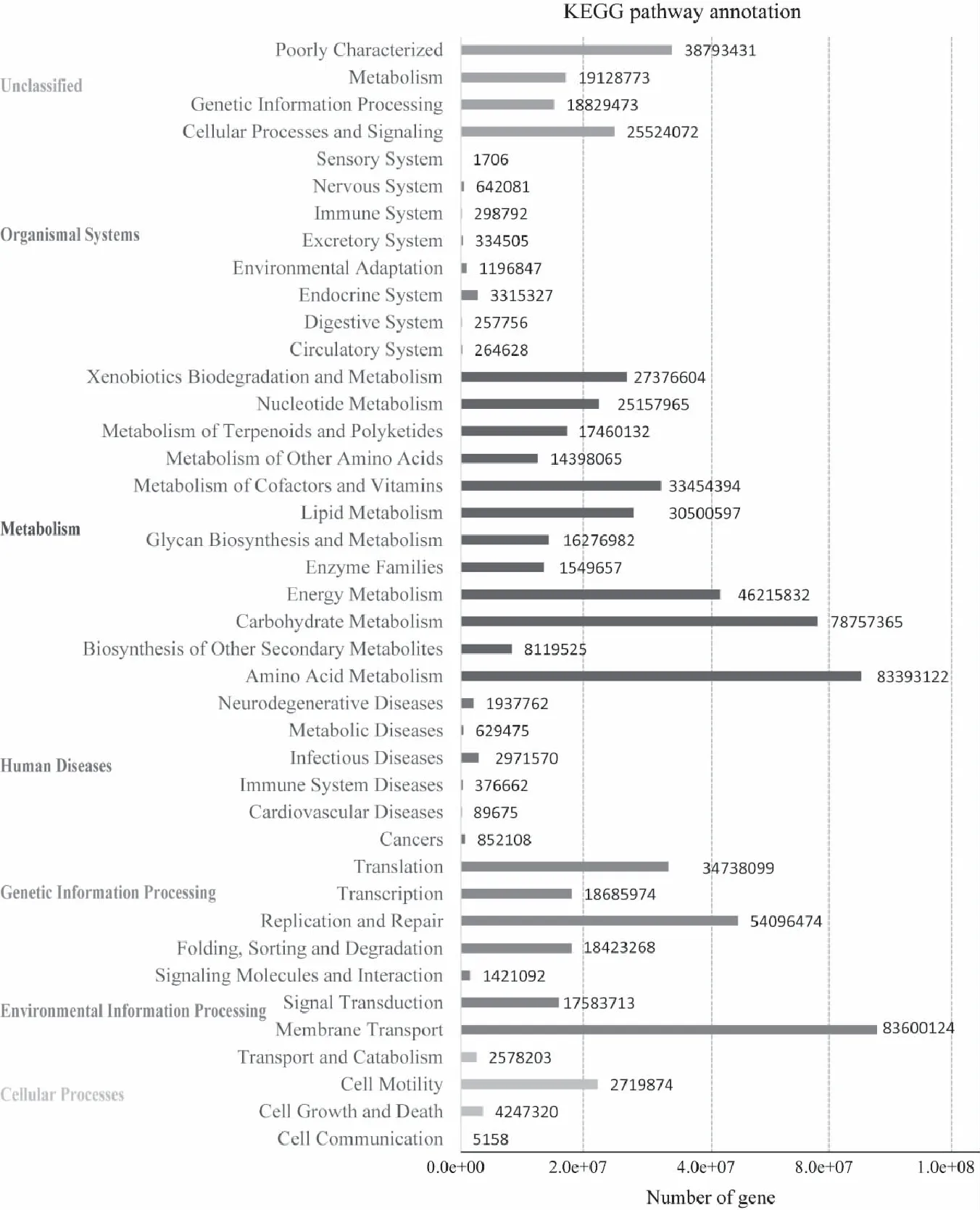
图5 预测功能基因Fig.5 Predicting functional genes
2.4.2 代谢通路与土壤性质的相关性分析
在探究了土壤微生物群落与土壤性质的相关性之后,本研究进一步对代谢通路与土壤性质的相关性进行了分析。结果表明(见图6):在白云岩喀斯特土壤中,土壤微生物代谢通路与土壤性质显著相关。土壤中的ECa2+、SOC、STP、TK、pH以及Emg2+都与代谢通路、基因信息处理、环境信息处理、细胞进程4个方面存在相关性。尤其以与代谢通路之间的相关性最为显著。其中,Emg2+对代谢通路的影响最为广泛,同时代谢通路与Emg2+的相关性也最为显著。包括43条与Emg2+呈正相关的代谢通路(31条显著正相关,如精氨酸和脯氨酸代谢、D-丙氨酸代谢、丙酮酸代谢等与之的相关性);13条代谢通路与之呈负相关关系(2条显著负相关)。表明在白云岩喀斯特土壤高镁的条件下,大量微生物代谢活动十分活跃。此外,pH作为土壤检测中的一项重要的理化指标,其与代谢通路仍有着密切的相关性。在本研究中,仅有苯丙素的生物合成(phenylpropanoid biosynthesis)、GPI-anchor生物合成等7条代谢通路与土壤pH呈负相关关系,其余52条代谢通路均与pH之间存在正相关关系。即随着pH值的增加,大量菌群的代谢活动也随之更加活跃。TK和STP与大多土壤微生物的代谢通路的相关性并不是很高。仅有类黄酮生物合成、青霉素和头孢菌素生物合成、半乳糖代谢等13条代谢通路与TK显著正相关。甜菜色素的生物合成、吲哚生物碱生物合成等6条代谢通路与STP呈显著负相关关系。较之代谢通路与pH和Emg2+的相关性,代谢通路与SOC和ECa2+之间主要存在负相关关系。存在18条与SOC显著负相关的代谢通路;代谢通路与ECa2+间基本呈负相关关系,仅有GPI-anchor生物合成和线粒体中脂肪酸链延长两通路与其正相关。而这两代谢通路与Emg2+间却是负相关。此外,丁酰苷菌毒与新霉素的生物合成、二苯乙烯类、庚酮类和姜酚的生物合成、青霉素和头孢菌素的生物合成、三羧酸循环、醚脂类代谢、嘧啶代谢6条代谢通路均与pH、Emg2+、TK、STP正相关,与SOC和ECa2+负相关。除此之外,Emg2+与环境信息处理、基因信息处理等通路之间的相关性也主要表现为正相关。说明在白云岩喀斯特地区,土壤高镁的条件下,土壤微生物在代谢、环境信息处理、基因信息处理等方面十分活跃。但对于其具体的作用机制,仍需进一步深入研究。

图6 土壤性质与代谢通路之间的相关性分析Fig.6 The relativity between soil property and metabolic pathway
3 讨论
土壤中微生物类群数量十分庞大,且土壤微生物的群落结构十分复杂。本研究中,相对丰度在1%以上的细菌门类有11个,以变形菌门(33.5%±2.8)、酸杆菌门(18.4%±1.5)和放线菌门(16.6%±1.9)丰度为最高。Liu等[18]基于Illumina平台,对樱桃果园中土壤微生物群落比较分析中显示,丰度最高的同为以上3个门类(变形菌门相对丰度为40.11%~63.72%,酸杆菌门相对丰度为4.02%~11.29%,放线菌门相对丰度为6.63%~13.00%)。此外,丰度在1%以上的还有TM7、厚壁菌门、疣微菌门、拟杆菌门和芽单胞菌门。与Liu等[18]相比较,本研究中的变形菌门丰度低于其樱桃果园土壤中变形菌门的丰度,而酸杆菌门和放线菌门则比其高。除TM7以外,其它几个主要的菌群门类丰度,都与本研究保持一致。此外,在Liu等[18]研究中,绿弯菌门、消化螺旋菌门以及浮霉菌门相对丰度都是低于1%,而在本研究中,以上3个菌群相对丰度则是高于1%。另外,将本研究中白云岩喀斯特土壤细菌群落与果园耕作土比较发现,2组之间的细菌群落丰度有的存在差异,有的丰度却接近。如:Duan等[19]对喀斯特石漠化地区桑树根际土壤微生物研究中,发现细菌中主要优势菌门为变形菌门、放线菌门和异常球菌-栖热菌门(deinococcus-Thermus),与本研究中的主要优势菌门为变形菌门、酸杆菌门、放线菌门存在一定的差异。可见,喀斯特石漠化地区桑树根际土壤细菌群落与喀斯特白云岩土壤细菌群落存在一定差异。这可能是植物根系对于根际微生物群落结构有影响,其根际微生物群落结构存在差异性[20]。为此,笔者推测地表植被与整个土壤微生物群落的分布相关。
高通量测序法的出现,所检测到的菌群数量远远超过预期的估计。戴雅婷等[21]基于高通量测序,对不同植被恢复类型下土壤微生物多样性进行了分析。牛世全等[22]基于IlluminaMiseq高通量测序技术对盐碱土壤微生物多样性进行了分析。Lauber等[23]首次利用高通量测序法将取自美国从北到南的88个土壤样品进行测序,同时基于宏观的角度,探究了细菌的群落结构组成,并发现细菌的群落组成多样性与其群落结构系统发育和土壤的pH存在显著的相关性,相关系数可达到0.79和0.71。本研究也证明了土壤微生物群落与pH之间存在相关性。此外,还有研究证明:土壤微生物生物量、微生物熵以及土壤基础呼吸作用均随着土壤pH值的降低而显著下降[24]。因此,在探究影响土壤微生物群落多样性的研究中,pH应作为重要环境因子而被关注。此外,本研究还发现,大部分微生物群落与Emg2+间呈负相关关系,说明微生物群落多样性的降低与喀斯特高镁的自然环境存在相关性,但目前还没有太多相关的研究来揭示其作用机理。
Picrust基因功能预测,作为一种新兴的研究手段,将不可视的微生物与KEGG数据库进行对比,进而预测基因功能。本研究表明,在白云岩喀斯特地区存在微生物携带大量与代谢有关的基因。可见,微生物的代谢活动较为活跃。此外,土壤环境因子与代谢通路间存在相关性。特别是Emg2+与代谢通路间的相关性尤为显著。但目前并没有太多与该研究方向相关的文献资料,因此,对其作用机制仍不明确,这也是后续研究亟待解决的问题。此外,PICRUSt基因功能预测在肠道微生物多样性研究中[25-27]也得到了广泛的应用,可作为将来一种常用的微生物研究方法。
本研究基于高通量测序数据,分析微生物群落结构,其数据深度能较好且全面的覆盖白云岩喀斯特土壤微生物,对于后续的白云岩喀斯特土壤微生物研究能提供数据支持。同时,基于Picrust算法预测获得的白云岩喀斯特土壤微生物功能基因组,进一步发现了土壤性质不仅与土壤微生物群落结构有关,与微生物群落的代谢也相关,可为白云岩喀斯特土壤微生物群落的代谢研究提供新的研究方向。
猜你喜欢
黑龙江水利科技(2020年8期)2021-01-21
中国比较医学杂志(2020年4期)2020-05-26
乡村地理(2019年2期)2019-11-16
石油地质与工程(2019年3期)2019-09-10
水生生物学报(2019年4期)2019-07-20
————水溶蚀岩石的奇观
家教世界(2019年4期)2019-02-26
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
录井工程(2017年4期)2017-03-16
文化月刊·下旬刊(2014年6期)2014-08-28
