
耐盐性谷氨酰胺酶在Bacillus subtilis 168 中的整合表达
2020-09-10 06:16徐朝阳徐美娟杨套伟饶志明
食品与生物技术学报 2020年6期
徐朝阳, 张 显, 徐美娟, 杨套伟, 饶志明*
(1. 江南大学 生物工程学院,江苏 无锡214122;2. 江南大学 工业生物技术教育部重点实验室, 江苏 无锡214122)
谷氨酰胺酶(EC 3.5.1.2)能将谷氨酰胺分解为谷氨酸和游离的氨气。 人体内的谷氨酰胺酶主要分为肾型谷氨酰胺酶和肝型谷氨酰胺酶[1],与细胞生长、代谢有关,可维持人体的酸碱平衡和内环境稳定。 谷氨酰胺酶还广泛存在于曲霉、酵母、细菌等微生物中。 其中, 在微生物中, 研究较多的有Micrococcus luteusK-3[2-3],Aspergillus oryzaeRIB40[4-5],Bacillussp LKG-01[6],Lactobacillus reuteriKCTC 3594[7]和Cryptococcus nodaensis[8]。 谷氨酰胺酶在日本酱油发酵过程中起着至关重要的作用,不仅可以增加酱油的风味,还可以增加酱油的营养成分。 但是,酱油酿造过程中的18%左右的高盐环境限制了谷氨酰胺酶的应用。 MaMoru wakayama 等人[2]研究表明,来源于Micrococcus luteusK-3 在8%~16%的盐浓度下可以稳定表达, 并且相对于无盐条件下,酶活提高了1.3 倍。所以,我们合成编码该谷氨酰胺酶的基因,将其在B. subtilis168 中表达。
枯草芽孢杆菌是一类非致病性细菌,被美国食品和药物管理局评定为安全菌株(GRAS)[9]。 食品工业在全球商业应用中,枯草芽孢杆菌被人类和动物当作益生菌使用[10],同时,它还能治疗胃肠道疾病[11]。 枯草芽孢杆菌是一个食品级表达宿主[12],可用于异源表达蛋白质和维生素[13]。
在微生物中,用质粒过表达外源基因时,在复制的过程中常出现不稳定的状态(ssDNA)[14],而导致质粒的丢失。 不添加抗生素条件下,会使这种现象很突出,从而限制了其在食品工业中的应用。 在之前的一项研究中,单拷贝β 半乳糖苷酶基因被整合到枯草芽孢杆菌的基因组DNA 中[15-16],重组菌株的酶活很低,我们可以通过提高外源基因的拷贝数来提高其表达量。 碱性蛋白酶E(aprE)和中性蛋白酶E(nprE)被认为是主要的蛋白酶,因为它们的活性在枯草芽孢杆菌分泌总蛋白酶活性的20%和70%[17-19]。 选用蛋白酶作为整合表达位点,既可以增加外源基因的拷贝数,又可以降低蛋白酶对外源蛋白的降解。 本研究将来源于Micrococcus luteusK-3的谷氨酰胺酶基因(Mglu)[2]以lox序列重组的方式定点整合入B. subtilis168 中的nprE 和aprE 基因位点,进而得到食品级表达谷氨酰胺酶的重组菌株。
1 材料与方法
1.1 菌株、质粒和引物
菌株、质粒、引物如表1 所示。

表1 菌株、质粒、引物一览表Table 1 Strains,plasmids and primers

续表1
1.2 培养基和试剂
LB 培 养 基(g/dL):蛋 白 胨1,酵 母 膏 0.5,NaCl 1。
发酵培养基(g/dL):葡萄糖2.5,安琪酵母4,NH4Cl 0.4,KH2PO40.1125,K2HPO4·3H2O 0.1875,CaCl20.2,L-谷氨酸钠0.2;pH 7.0。
SPI 培养基(g/dL):KH2PO40.6,K2HPO4·3H2O 1.83, (NH4)2SO40.2,Na3C6H5O7·2H2O 0.1,MgSO4·7H2O 0.02,葡萄糖0.5,酪蛋白水解物0.02,酵母提取物0.1,色氨酸0.005;115 ℃,灭菌25 min。
SPII 培养基:SPI 培养基体积分数98%,CaCl22.7 mg/L,MgCl20.48 mg/L;115 ℃,灭菌25 min。
100×EGTA:10 mmol/L EGTA 溶 液,NaOH 调pH 值至8.0。
质粒提取试剂盒、蛋白纯化试剂盒、基因组提取试剂盒、PCR 试剂:购于中国大连市TaKaRa 公司。
1.3 重组B. subtilis 168 的构建
1.3.1 B. subtilis 168 基因组的提取B. subtilis 168 基因组的提取按照基因组提取试剂盒操作手册进行。
1.3.2 整合片段同源臂的扩增通过NCBI 查询可得到B. subtilis 168 aprE、nprE 基因的序列,选择两侧大约600~700 bp 的片段作为同源臂,设计引物扩增同源臂。 同源臂PCR(聚合酶链式反应)体系中,模板为B. subtilis 168 基因组,aprE 基因上下游同源臂引物对分别是P1、P2 和P7、P8,nprE 基因上下游同源臂引物为P9、P10 和P15、P16。 PCR 扩增产物核酸胶回收后,用胶回收试剂盒纯化并测定核酸浓度。
1.3.3 lox71-zeo-lox66 基因片段的扩增通过NCBI 网站查询到p7Z6 质粒全基因序列,根据其中lox71-zeo-lox66 的基因序列, 设计引物扩增lox71-zeo-lox66。 PCR 体系中,模板为p7Z6,aprE 基因位点lox71-zeo-lox66 基因的引物为P3 和P4,nprE 基因位点lox71-zeo-lox66 基因的引物为P11 和P12。PCR 扩增产物核酸胶回收后,用胶回收试剂盒纯化并测定核酸浓度。
1.3.4 HpaII-Mglu 基因片段的扩增根据HpaII和Mglu 基因的序列设计引物对扩增HpaII-Mglu 基因,PCR 体系中,模板为pMA5-HpaII-Mglu,aprE 基因插入位点HpaII-Mglu 基因的扩增引物为P5 和P6,nprE 基因插入位点HpaII-Mglu 基因的扩增引物为P13 和P14。PCR 扩增产物核酸胶回收后,用胶回收试剂盒纯化并测定核酸浓度。
1.3.5 上下游同源臂、lox71-zeo-lox66 基因、HpaIIMglu 基因的融合4个基因片段的融合,参照两步法融合方法[21],操作具体如下:
第1 步PCR 体系如表2 所示:
PCR 条件:98 ℃预变性3 min;98 ℃解链8 s,61 ℃退火5 s,72 ℃延伸4 min,共13个循环;72 ℃保温20 min。

表2 4 片段融合第1 步PCR 体系Table 2 First step PCR fusion consists of four segments
第2 步PCR 体系如表3 所示:
PCR 条件:98 ℃预变性3 min;98 ℃解链10 s,61 ℃退火15 s,72 ℃延伸4 min, 共28个循环;72 ℃保温20 min。
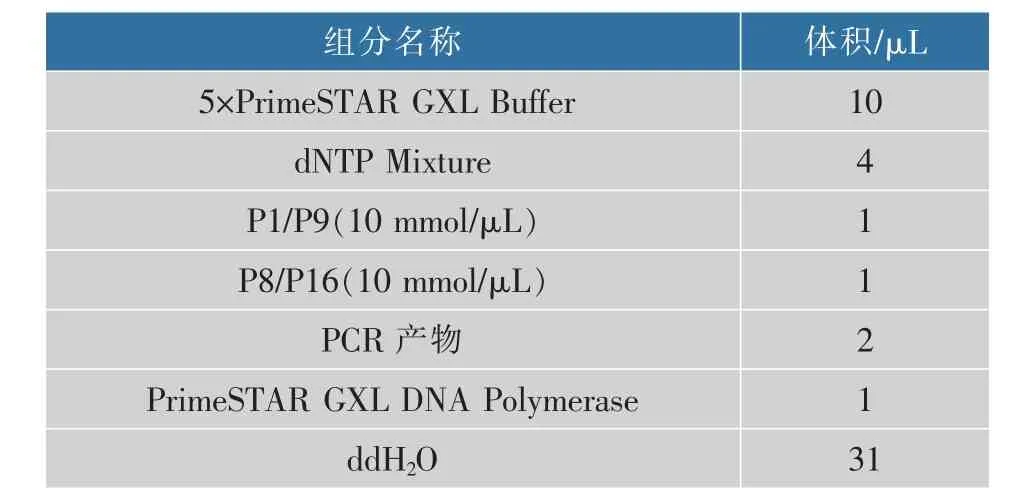
表3 4 片段融合第2 步PCR 体系Table 3 Second step PCR confusion consists of four segments
1.3.6 B. subtilis 168 感受态制备及转化将-40 ℃甘油管保存的B. subtilis168 在LB 固体琼脂平板上划线活化,37 ℃恒温培养箱中培养12 h, 取单菌落接种于10 mL LB 液体培养基中,在37 ℃、180 r/min培养12 h。转接200 μL 菌液于SPI 培养基中,37 ℃、180 r/min 培养5 h, 转接1 mL 菌液于SPII 培养基中,37 ℃、180 r/min 培养1.5 h。 接着,于SPII 培养基中加入110 μL 100×EGTA,37 ℃、180 r/min 培养10 min。 所得菌液即为B. subtilis168 感受态细胞,将菌液分装,500 μL 每管。转化时加入重组DNA 片段, 轻轻振荡混匀。 37 ℃、180 r/min 条件下培养90 min,涂布含有博莱霉素(25 mg/L)的LB 固体琼脂平板,置于37 ℃恒温培养箱中倒置培养12 h[22]。
1.3.7 抗性基因的敲除pTSC 质粒表达CRE 酶,会引导lox序列重组,消除抗性基因。 将上述方法得到的B. subtilis168 转化子,再次制作成感受态。 将pTSC 质粒转化入感受态细胞中,涂布到含有卡那霉素(100 mg/L)的固体LB 平板,37 ℃恒温培养箱培养12 h。 将得到含有阳性转化子在51 ℃恒温培养箱中培养24 h,使温度敏感型质粒pTSC 丢失。
1.3.8 重组B. subtilis 168 的构建按照上述方法制作B. subtilis168 感受态之后, 将得到的aprE、nprE 位点的融合片段分别转化到B. subtilis168 感受态中,筛选到阳性转化子,按照上述方法将抗性基因敲除后得到重组菌株BSM1 和BSM2。 接着,在BSM1 的基础上, 将nprE 位点的融合片段转化到BSM1 的感受态中,筛选到阳性转化子,敲除抗性基因,得到BSM3。
1.4 重组B. subtilis 168 的发酵
将重组菌株分别在LB 平板上划线, 挑选单菌落接种于10 mL LB 培养基中,37 ℃、180 r/min 培养12 h。将1 mL 菌液转接入100 mL 发酵培养基中(500 mL 锥形瓶),于24 ℃、180 r/min 培养24 h。 收集得到的菌体用于酶活测定和SDS-PAGE 分析。
1.5 重组谷氨酰胺酶的蛋白纯化及SDS-PAGE 分析
1.5.1 粗酶液的制取将发酵得到的菌体,10000 r/min 离心5 min 后收集菌体,用50 mmol/L 的Tris-HCl(pH 7.5)缓冲液洗涤2次后,用破碎缓冲液悬浮菌体。 加入溶菌酶 (终质量浓度为1 mg/mL),混匀,在4 ℃条件下放置2 h。 将离心管置于冰中,超声破碎细胞,条件为:功率400 W,工作1 s 停2 s,总时间为5 min。 将破碎后的菌液,在4 ℃条件下,10000 r/min 离心20 min,获得上清液。 将上清液用0.45 μm 滤膜过滤,即可得到粗酶液。
1.5.2 蛋白质纯化将粗酶液用Ni-NTA 蛋白纯化柱进行纯化。 纯化操作按照AKTA 蛋白纯化仪说明书进行。上样体积为5 mL,洗脱流量为0.5 mL/min。
1.5.3 谷氨酰胺酶的SDS-PAGE 分析分别吸取80 μL BSM3 重组菌株的纯酶液、 粗酶液和B.subtilis168 的粗酶液,加入20 μL 5×Loading Buffer,混匀后煮沸20 min,进行SDS-PAGE 分析。 分离胶浓缩胶体积分数分别为12%和4%。 上样量为15 μL。
1.6 酶活测定
本研究使用的是通过测定产物谷氨酸的含量[23],进而测定谷氨酰胺酶酶活的方法。 具体如下:
酶活测定体系(1 mL)为:850 μL 用50 mmol/L Tris-HCl(pH 7.5)溶解50 mmol/L 谷氨酰胺的底物溶液,37 ℃水浴锅预热5 min, 加入20 μL 粗酶液,对照加入100 μL 15 g/dL 的三氯乙酸,混匀后在37 ℃恒温水浴锅反应5 min, 迅速加入100 μL 15 g/dL的三氯乙酸并混匀, 对照加入20 μL 粗酶液并混匀,反应终止。 将反应液10000 r/min 离心10 min,以去除蛋白质,接着,取25 μL 反应液测定谷氨酸含量。 酶活定义:在37 ℃条件下,1 min 内反应生成1 μmol 的谷氨酸所需的酶量定义为一个酶活单位。
1.7 重组菌株的稳定性分析
单细胞微生物的生长繁殖,细胞呈现几何级数2n增长,当n=7 时,27=128,将静止期的菌液按体积分数1%接种量接种,再将其培养至静止期,即可认为细胞已连续传代7次[24-25]。 分别将BSM、BSM3 重组菌株在LB 琼脂平板上划线,37 ℃恒温培养箱培养12 h。 接着,分别挑选BSM、BSM3 重组菌株单菌落接种至LB 液体培养基中,37 ℃、180 r/min 条件下培养12 h,作为种子液。分别将1 mL 菌液转接至100 mL 发酵培养基(500 mL 锥形瓶),24 ℃、180 r/min条件下培养24 h 后,分别取样测酶活,同时继续将1 mL 菌液转接至100 mL 新鲜发酵培养基(500 mL锥形瓶)。 依此类推,每隔24 h 取样测定酶活,共测定6次。
1.8 重组菌BSM35 L 发酵罐发酵水平分析
将重组菌株BSM3 在LB 固体平板划线,37 ℃恒温培养箱培养12 h。挑取单菌落接种至10 mL LB培养基中,37 ℃、180 r/min 条件下培养12 h。 吸取1 mL 菌液转接于100 mL LB 培养基中 (1000 mL锥形瓶),37 ℃、180 r/min 条件下培养10 h,作为种子液。 接着将种子液全部转接进5 L 发酵罐中(2 L发酵培养基),控制条件:温度为24 ℃,pH 为7.0(体积分数50%磷酸和50%氨水控制)、转速为450 r/min。
2 结果与讨论
2.1 重组菌株构建
重组菌株构建过程,见图1,我们根据上述方法扩增出了各个片段(见图2),并构建了2个蛋白酶基因位点的融合片段 (见图3), 并成功转化入B.subtilis168 感受态中,通过博莱霉素抗生素平板筛选到阳性转化子,接着用pTSC 质粒去除抗性基因,成功得到无抗性基因的重组菌株BSM1 和BSM2。在重组菌株BSM1 的基础上,在nprE 基因位点插入Mglu基因,去除抗性基因后,得到重组菌株BSM3。通过增加谷氨酰胺酶基因Mglu的整合表达位点,可能能解决单拷贝基因表达产物低的问题。

图1 Mglu 基因整合入B. subtilis 168 染色体DNA 过程示意图Fig. 1 Strategy for integrating Mglu gene in B. subtilis 168 chromosome DNA
一个融合片段包括两段同源臂、HpaII-Mglu基因和lox71-zeo-lox66 基因。 由于同源区域的存在,B. subtilis168 染色体DNA 会在敲除基因(aprE 或nprE)的同时,引入HpaII-Mglu基因和lox71-zeolox66 基因。 接着,会通过CRE/loxp系统敲除lox71序列和lox66 序列之间的zeo抗性基因, 并形成了lox72 序列。

图2 lox71-zeo-lox66、HpaII-Mglu 和上下游同源臂片段的PCR 电泳图Fig. 2 PCR electrophoresis of lox71-zeo-lox66,HpaIIMglu and two homologous arm segments
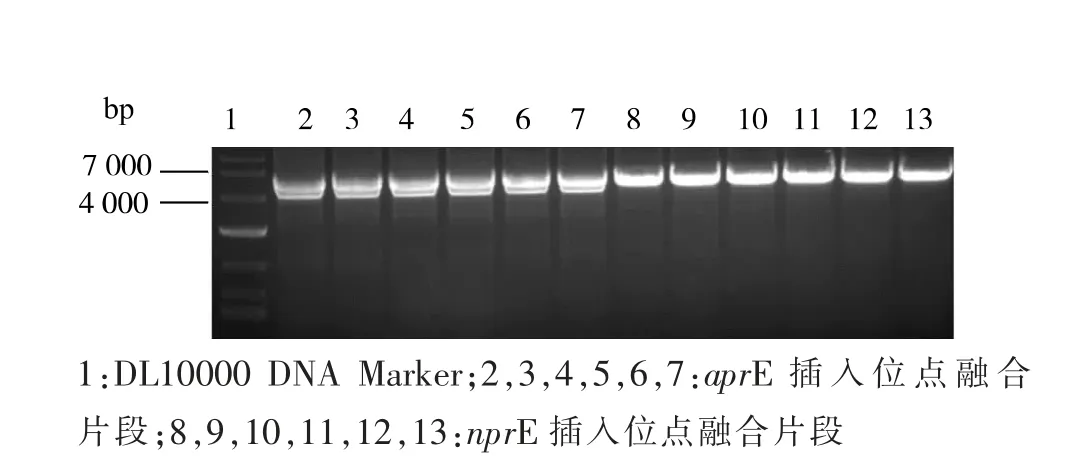
图3 融合片段PCR 电泳图Fig. 3 PCR electrophoresis of fusion segaments
2.2 重组菌株的发酵酶活对比
为了探究不同重组菌株的谷氨酰胺酶表达水平, 按照上述方法对菌株B. subtilis 168、BSM1、BSM2、BSM3 进行发酵培养。 如图4 所示,重组菌株的生长趋势和野生型菌株B. subtilis 168 趋势基本保持一致, 在18 h 时达到最大菌浓,OD600为9 左右。 重组菌株的最大发酵酶活如图5 所示,野生型菌株B. subtilis 168 无谷氨酰胺酶酶活, 重组菌株BSM1、BSM2 和BSM3 的最大发酵酶活分别为0.5、0.6 U/mL 和1.0 U/mL。 重组菌株BSM1 和BSM2 中Mglu 基因的拷贝数都是1, 但BSM2 相对于BSM1的谷氨酰胺酶酶活要高,原因可能是中性蛋白酶对表达的谷氨酰胺酶降解量更多,将其敲除之后更有利于谷氨酰胺酶的表达。很显然,重组菌株BSM3 的发酵酶活最高, 重组菌株BSM3 相对于BSM1 酶活提高了100%(图5)。使用整合表达外源基因可以避免游离质粒表达分离不稳定的问题,但是目的基因表达量低成为整合表达的一大障碍。 我们可以通过增加整合表达位点和敲除内源性蛋白酶的方法增加目的蛋白基因的表达量。

图4 重组菌株生长曲线Fig. 4 Growth curve of recombinant strains
2.3 重组谷氨酰胺酶的蛋白质纯化
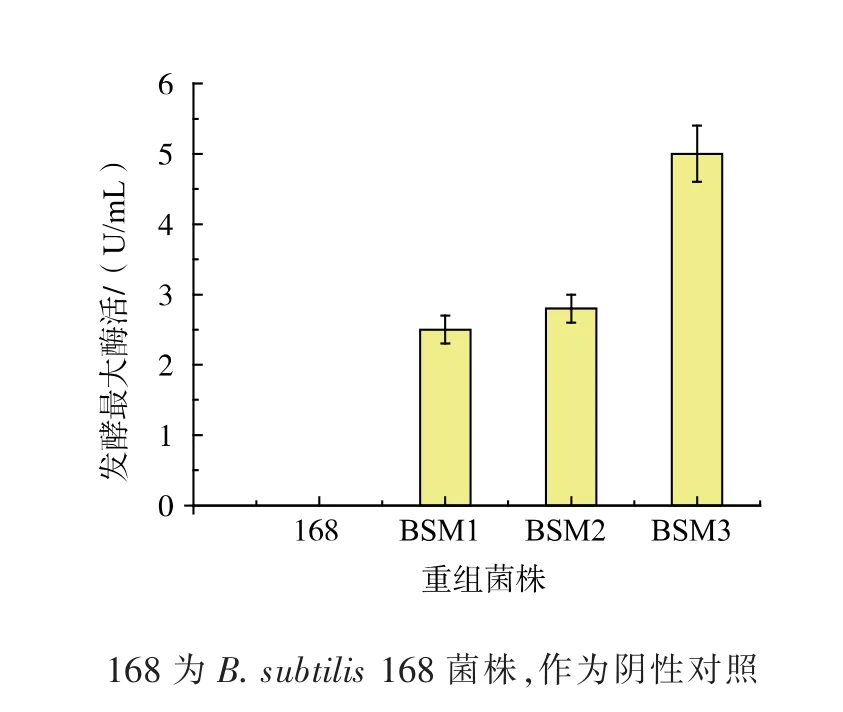
图5 重组菌株和B. subtilis 168 最大发酵酶活对比分析Fig. 5 Maximum fermentation glutaminase activities of the recombinant strains and B. subtilis 168
在重组菌株构建过程中,Mglu 基因前面3’端添加了His-tag 序列,所以可以通过Ni2+亲和层析分离出谷氨酰胺酶。 纯化后的谷氨酰胺酶的SDSPAGE 电泳图如图6 所示,以B. subtilis 168 作为阴性对照, 粗酶液和纯酶液在大约4.8×104位置有一条明显的条带。 酶活测定可知,BSM3 表达的谷氨酰胺酶比酶活为1326 U/mg,大小基本与报道一致[2]。

图6 重组菌株BSM3 表达谷氨酰胺酶SDS-PAGE 电泳图Fig. 6 SDS -PAGE analysis of the expression of glutaminase in BSM3
2.4 重组菌株BSM3 的遗传稳定性分析
整合型菌株的遗传稳定性是衡量工业生产菌株的一个重要因素。 相对于游离质粒表达,整合表达可以提高外源基因的遗传稳定性。 在无抗生素条件下, 我们通过连续转接重组菌株并测定酶活,分析重组菌株的遗传稳定性,结果如图7 所示,重组菌株BSM3 在连续传代42次之后, 酶活依然稳定,遗传稳定性较好。 然而,游离质粒表达Mglu 基因重组菌株BSM 在连续传代14次之后,酶活明显下降,遗传稳定性很差。 微生物使用游离质粒过表达谷氨酰胺酶的优势很明显,可以大大地提高酶活。 但是,质粒的分离稳定性很差,被认为是其表达的一大问题,限制了在工业中的应用。 所以,整合型表达谷氨酰胺酶菌株BSM3 更适合工业生产。

图7 重组菌株BSM3、BSM 遗传稳定性分析Fig. 7 Analysis of genetic stability of the recombinant strains BSM3 and BSM
2.5 5 L 发酵罐的分批补料发酵
由于摇瓶发酵营养物质有限,限制了重组菌株BSM3 的生物量,为了提高谷氨酰胺酶的产量,我们选择使用5 L 发酵罐进行分批补料发酵。 结果如图8 所示,重组菌株BSM3 在7 h 左右时进入对数期。在32 h 左右时,OD600达到了最大,约为92。 我们发现,产酶曲线和生长曲线存在一定的相关性。 酶活在32 h 左右是达到了最大,为41.5 U/mL。 32 h 之后,谷氨酰胺酶酶活有逐渐降低的趋势,这可能是因为该谷氨酰胺酶不稳定,发生了变性,导致酶活降低。

图8 重组菌株BSM35 L 发酵罐发酵水平分析Fig. 8 Fed-batch fermentation of the recombinant strain BSM3
3 结 语
本实验构建稳定的重组谷氨酰胺酶表达菌株,选择对B. subtilis 168 进行定点整合,来表达谷氨酰胺酶,提高Mglu 基因的遗传稳定性。 增加基因表达产物活性可以通过在基因上游插入强启动子、增加结构基因拷贝数、对结构基因进行改造、降低降解或抑制表达产物活性物质的表达等方式进行。 本实验中在使用强启动子HpaII 的基础上, 在敲除2个内源性蛋白酶基因aprE、nprE 的同时, 插入谷氨酰胺酶基因。 既增加了谷氨酰胺酶的拷贝数,又降低了蛋白酶对表达产物谷氨酰胺酶的降解。 通过对比发现,增加谷氨酰胺酶基因拷贝数,可有效提高谷氨酰胺酶表达水平。 本实验中采用lox 序列重组的方式将Mglu 基因整合到B. subtilis 168 染色体上,抗性基因也插入到了染色体上, 表达pTSC 质粒上重组酶CRE,引导lox 序列重组,消除抗性基因,因此, 该重组表达谷氨酰胺酶菌株BSM3 可直接用于食品发酵。
猜你喜欢
现代妇产科进展(2022年9期)2022-10-07
中国饲料(2022年5期)2022-04-26
中国土壤与肥料(2021年5期)2021-12-02
天津医科大学学报(2021年2期)2021-03-29
岭南现代临床外科(2020年5期)2020-12-13
农产品加工(2020年17期)2020-10-22
疯狂英语·新悦读(2020年7期)2020-07-30
食品安全导刊·中旬刊(2020年2期)2020-06-01
癌变·畸变·突变(2020年1期)2020-02-12
文苑(2018年22期)2018-11-19
