
用非模型依赖法和威布尔模型法研究富马酸替诺福韦酯口服固体制剂的溶出行为
2020-09-02 06:55秦淑娜顾圣莹汪硕闻陈永智范国荣
药学服务与研究 2020年4期
秦淑娜,顾圣莹,汪硕闻,陈永智,范国荣*,范 琦*
(1.重庆医科大学药学院药物分析教研室,重庆 400016;2.上海交通大学附属第一人民医院临床药学科,上海 200080)
富马酸替诺福韦酯(tenofovir disoproxil fumarate, TDF)是一种核苷酸逆转录酶抑制剂前药,可在细胞内磷酸化为活性代谢产物替诺福韦双磷酸盐,其通过竞争性抑制病毒的逆转录酶,终止DNA链增长,主要用于治疗人免疫缺陷病毒(human immunodeficiency virus,HIV)及乙型肝炎病毒(hepatitis B virus,HBV)感染[1-2]。《国家免费艾滋病抗病毒药物治疗手册(第四版)》中,将TDF列为一线治疗药物。TDF片由美国Gilead Sciences公司研发,于2003年获得美国食品药品监督管理局(FDA)批准,以商品名韦瑞德(Viread)在美国上市,用于治疗HIV感染。TDF片于2008年获得欧盟和FDA批准用于治疗乙型肝炎;2008年6月该药获得中国国家食品药品监督管理局的进口药品注册证,专利于2017年到期后,国产仿制药获批上市。
TDF为生物药剂学分类系统(biopharmaceutics classification system,BCS)Ⅲ类药物,具有高溶解性和低渗透性。《中华人民共和国药典》(简称《中国药典》)2015年版中未收录该药,国内文献关于该药的溶出度一致性评价研究也较少。因此建立可靠、稳定的TDF口服固体制剂溶出度测定方法,可为国内TDF制剂质量标准的建立提供参考依据。
溶出度试验模拟口服固体制剂在人体消化道内崩解和溶出过程,是评价口服固体制剂质量的重要手段之一[3-4]。本研究建立TDF口服固体制剂溶出度测定的HPLC法,并对国内4个厂家生产的TDF仿制药进行溶出度一致性评价,为临床遴选优质的仿制药提供参考,并为国产TDF制剂质量标准的建立提供数据支持。
1 仪器和试药
1.1 仪器 CPA225D分析天平(精度0.01 mg,德国赛多利斯公司);UltiMate 3000高效液相色谱仪(美国赛默飞公司);Waters C18色谱柱(250 mm×4.6 mm,5 μm,美国Waters公司);FE 20 pH计(梅特勒-托利多国际有限公司);SNTR-6400A溶出度仪、SSAS-6000自动取样器(日本岛津公司);MCE针头式过滤器(0.45 μm,上海岛津技迩商贸有限公司);5804R高速冷冻离心机(德国艾本德公司);SK5200H超声波清洗器(上海科导超声仪器有限公司);全自动渗透纯水机(上海金迈净水设备有限公司)。
1.2 试剂和药品 TDF对照品(含量>98%,批号M0402AS,大连美仑生物技术有限公司);TDF药品信息见表1。乙腈(色谱纯,批号JA060230,默克股份两合公司);盐酸(批号20171108)、磷酸(批号20160217)、乙酸(批号201709012)、甲酸(批号20160620)、氢氧化钠(批号20160620)、无水乙酸钠(批号20170908)、磷酸二氢钠(批号20160505)及磷酸二氢钾(批号20160325)均为分析纯,购于上海凌峰化学试剂有限公司;实验用水均由上海交通大学附属第一人民医院临床药学科利用全自动渗透纯水机自制。
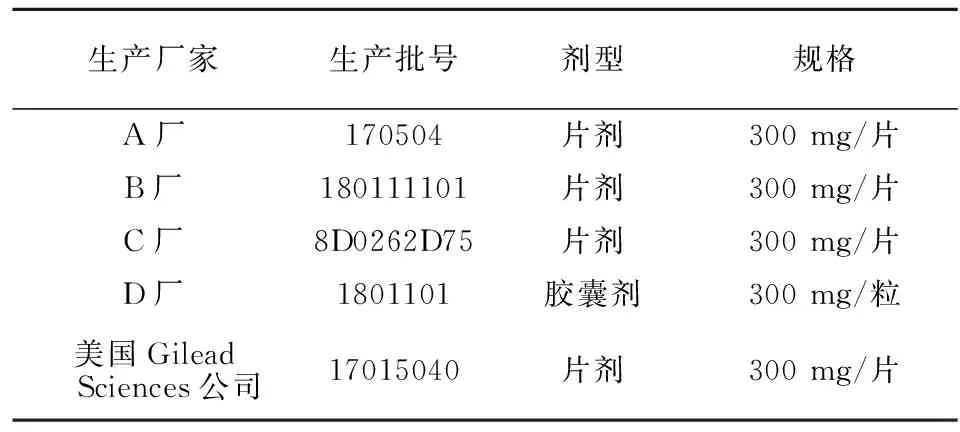
表1 富马酸替诺福韦酯口服固体制剂的药品信息Table 1 The drug information of the oral solid preparations of tenofovir disoproxil fumarate
2 方法和结果
2.1 溶出条件 参照《中国药典》2015年版四部通则0931选择桨法,桨转速为50 r/min。根据《普通口服固体制剂溶出度试验技术指导原则》(简称《溶出度指导原则》),采用4种不同pH值的溶出介质,溶出介质体积为900 ml(由于D厂生产的TDF口服固体制剂为胶囊剂,根据《中国药典》2015年版四部通则0931以桨法实验时需用沉降篮),温度为(37±0.5) ℃,取样时间点为5、10、15、20、30 min,各取样时间点取溶出液5 ml,并及时补充相应同温度同体积的溶出介质。
2.2 溶液的配制
2.2.1 溶出介质 4种溶出介质分别为:(1)纯化水;(2)pH=1.2的盐酸溶液;(3)pH=4.0的缓冲溶液;(4)pH=6.8的缓冲溶液。配制方法参考《溶出度指导原则》。
2.2.2 对照品溶液 取TDF对照品约10 mg,精密称定,置10 ml容量瓶中,加80%甲醇水溶液稀释至刻度,得TDF对照品储备液,再将对照品储备液用4种溶出介质分别稀释至相应浓度,即得TDF对照品溶液。
2.2.3 供试品溶出液 取TDF片剂6片或胶囊剂6粒,置于2.2.1项下配制的溶出介质中,按2.1项下条件进行溶出度试验,得溶出液,置于冷冻离心机中,1.803 2×104×g离心10 min,取上清,即得。
2.3 测定方法的建立和验证
2.3.1 色谱条件 以含0.1%甲酸的乙腈为流动相A,0.02 mol/L磷酸氢二钠水溶液为流动相B,梯度洗脱(0~6 min,75%~55% B;6~9 min,75% B);检测波长260 nm;流速1 ml/min;柱温30 ℃;进样量10 μl。
2.3.2 专属性考察 分别取2.2项下4种空白溶出介质、4种对照品溶液及TDF原研药溶出液(取样时间点为30 min),1.803 2×104×g离心10 min后,按2.3.1项下色谱条件测定,结果4种空白溶出介质在主峰位置无干扰,不同溶出介质中替诺福韦酯峰的保留时间基本一致,对照品溶液与原研药溶出液中的替诺福韦酯峰保留时间稳定在8.0 min左右(见图1),表明该方法专属性较好。
2.3.3 线性关系考察 精密量取2.2.2项下对照品储备液适量,分别用4种溶出介质稀释成103.01、206.02、309.03、515.05、721.07、927.09、1 030.10 μg/ml系列浓度对照品溶液,1.803 2×104×g离心10 min后,按2.3.1项下色谱条件测定。以浓度(c)为横坐标,峰面积(A)为纵坐标进行线性回归,回归方程见表2。结果显示,4种溶出介质中,TDF在103.01~1 030.10 μg/ml浓度范围内与峰面积具有良好的线性关系,且截距均不超过100%溶出时峰面积的2%,表明系统误差较小[4]。
2.3.4 精密度实验 精密量取2.2.2项下TDF对照品储备液适量,分别用4种溶出介质稀释成343.37 μg/ml(相当于100%浓度水平)的对照品溶液,1.803 2×104×g离心10 min后,按2.3.1项下色谱条件测定,连续进样6次,结果显示,平均峰面积的RSD均<2%(见表2),表明日内精密度符合要求。以上述用4种溶出介质为稀释液,分别平行配制6份对照品溶液,1.803 2×104×g离心10 min后,按2.3.1项下色谱条件测定,连续进样6次,计算峰面积的RSD;连续测定3 d,计算平均峰面积的RSD。结果显示,平均峰面积RSD均<2%(见表2),表明方法重复性及日间精密度均符合要求。
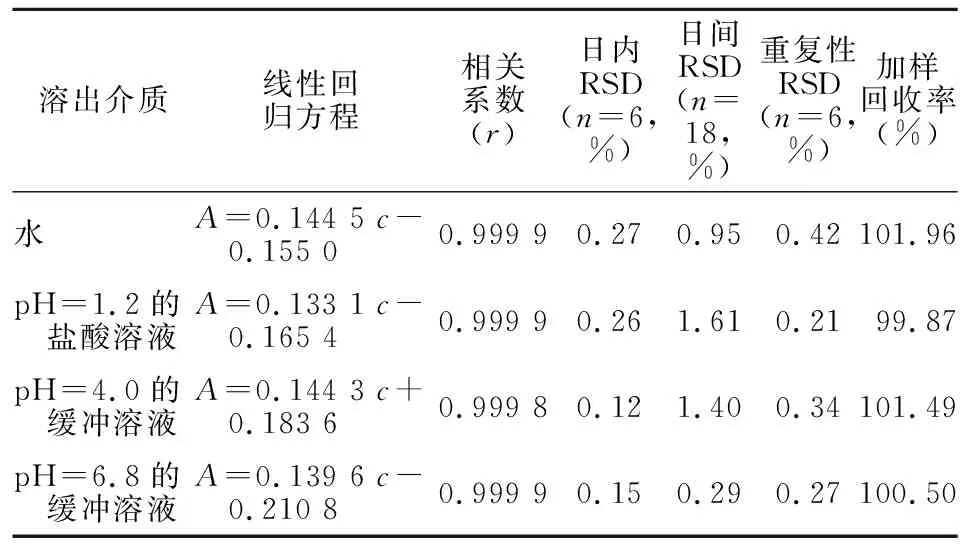
表2 HPLC法测定TDF浓度的方法学验证Table 2 Validation of HPLC method for concentration determination of TDF
2.3.5 回收率实验 取TDF原研药按2.2.3项下方法操作,取30 min时的溶出液适量,其余溶出条件同2.1项。将相当于制剂标示量100%的对照品加入溶出液中,平行配制6份,将加样前后溶液以1.803 2×104×g离心10 min后,按2.3.1项下色谱条件测定,根据两次测定结果计算加样回收率。结果显示,平均加样回收率在99%~102%(见表2),表明该方法准确度符合要求。
2.3.6 稳定性实验 精密量取2.2.2项下TDF对照品储备液适量,分别用4种溶出介质稀释,平行制备6份浓度为343.37 μg/ml(相当于100%浓度水平)的对照品溶液。分别于37 ℃水浴放置0、3 h及室温放置0、3、6、12 h 后,1.803 2×104×g离心10 min,按2.3.1项下色谱条件测定,计算峰面积平均值。结果显示峰面积平均值的RSD<2%(n=6),表明在4种溶出介质中,TDF于37 ℃放置3 h及室温下放置12 h均稳定。
2.4 桨转速的选择 将溶出仪桨转速分别设为50和75 r/min,取TDF原研药和A厂仿制药片各6片,以水为溶出介质,按2.2.3项下方法制备供试品溶出液,然后按2.3.1项下色谱条件测定,计算平均溶出度百分比。结果显示,桨转速为75 r/min时TDF仿制药与原研药溶出均较快,其中A厂生产的仿制药片在第一个取样时间点(5 min)时基本全部溶出,溶出度为(98.80±2.35)%(n=6),50 r/min时TDF仿制药与原研药溶出区分力度更大,故将50 r/min作为本研究的桨转速。
2.5 溶出度条件耐用性考察
2.5.1 溶出介质温度、桨转速耐用性考察及溶出介质脱气的影响 分别取TDF原研药6片,以水为溶出介质,按2.2.3项下方法制备供试品溶出液,在给定的溶出条件下分别考察溶出介质温度(37 ℃、36 ℃、38 ℃)和桨转速(50、45、55 r/min)及溶出介质是否脱气对溶出的影响。分别对条件改变前后的溶出曲线做t检验,结果P值均>0.05,表明溶出介质微小温度变化、桨转速变化及溶出介质是否脱气对TDF口服固体制剂的溶出行为无显著影响。
2.5.2 滤膜吸附影响的考察 取TDF原研药,按2.2.3项下方法分别制备溶出液,仅于溶出30 min时取样,分成两份,一份用MCE针头式过滤器过滤,另一份于1.803 2×104×g离心10 min后取上清液,分别平行制备6份,按2.3.1项下色谱条件测定。结果在水、pH=1.2的盐酸溶液、pH=4.0的缓冲溶液和pH=6.8的缓冲溶液中,滤膜过滤样品与离心后样品的峰面积平均值的百分比分别为93.07%、95.81%、94.68%及94.38%,即在4种溶出介质中,TDF滤膜吸附率均>2%。为避免滤膜对供试品溶出液中药物的吸附作用,本研究进样前采用离心的方法处理溶出液。
2.6 溶出度一致性评价
2.6.1 非模型依赖法 按2.2.3项下方法制备TDF口服固体制剂仿制药及原研药溶出液,然后按2.3.1项下色谱条件进样,计算各取样时间点的累积溶出百分率。根据《溶出度指导原则》,绘制溶出曲线,见图2。由于D厂生产的TDF胶囊剂仅在pH=1.2的盐酸溶出介质中1 h内可以全部溶出,故将D厂生产的TDF胶囊剂在其他3种溶出介质中的取样时间点设为5、10、15、20、30、45、60、75、90、120、150、180 min,其余溶出条件同2.1项,计算各取样时间点的累积溶出百分率并绘制溶出曲线(见图3)。结果除D厂生产的TDF胶囊剂外,其余仿制药片剂与原研药在15 min内的溶出度均>85%。根据《溶出度指导原则》,无需比较相似因子f2,即可认为A、B、C厂生产的仿制药片剂与原研药的溶出行为一致,D厂生产的TDF胶囊剂与原研药溶出行为不一致。
2.6.2 威布尔模型法 威布尔模型法可以精确描述药品溶出过程[5-6],故本研究采用威布尔模型法对TDF固体制剂的溶出行为进行评价。由于TDF片剂在溶出介质中崩解溶出极快,设位置参数α为0,再将3种国产TDF片剂和原研药溶出度试验所得数据按威布尔模型法采用Excel软件(Office 2016)建模,并计算4种溶出介质中各释放参数的均值[6-7](见表3)。结果显示,不同厂家生产的TDF片剂在4种溶出介质中药物溶出50%的时间参数(T50)、药物溶出63.2%的时间参数(Td)和药物溶出85%的时间参数(T85)均值均不相同,表明A、B、C厂生产的仿制药片剂与原研药的溶出参数存在差异,其中C厂生产的TDF片剂在4种溶出介质中的T50、Td和T85分别是A厂的35.31、18.08及5.21倍,说明国内不同厂家生产的TDF片剂的溶出行为存在较大差异。
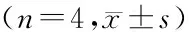
表3 TDF片的溶出参数Table 3 Dissolution parameters of TDF tablets
3 讨 论
本研究建立了TDF浓度测定的HPLC法,方法的专属性、准确度、精密度、回收率及稳定性均符合要求。本研究参考《溶出度指导原则》,采用4种不同溶出介质,溶出度条件耐用性考察结果符合要求,故本研究建立的方法适用于TDF口服固体制剂溶出行为的一致性评价。
本研究发现,D厂生产的胶囊剂与原研药溶出行为不一致,且在不同溶出介质中胶囊内药物的溶出行为也不同。可能是因为D厂生产的TDF口服固体制剂剂型改变所致,明胶外壳对TDF药物的溶出有限制。有文献指出,TDF胶囊剂和片剂具有生物等效性[8],可能与其生物药剂学特性有关。另外,由于TDF片剂中主药释放速度较快,可产生突释效应,导致胃肠道不良反应等,而TDF胶囊剂的释放速度较平缓,可避免突释效应,此现象可为临床选药提供参考。
本研究采用两种方法对TDF原研药与国产口服固体制剂溶出度的一致性进行评价。首先,参照《溶出度指导原则》绘制溶出曲线,其中除D厂生产的胶囊剂外,其余3个厂家生产的仿制药片剂与原研药在4种溶出介质中15 min内溶出度均>85%,根据《溶出度指导原则》,理论上初略表征3种片剂仿制药与原研药的溶出行为一致。将这3个厂家生产的TDF片剂与原研药以威布尔模型法计算溶出参数,结果3种TDF片剂与原研药之间,及国产仿制药片剂之间的溶出参数存在差异,由此推测国内不同厂家生产的TDF片剂的生产工艺可能存在较大差异。对溶解较快的药品,仅以15 min溶出量>85%即认为口服固体制剂的溶出行为一致具有局限性,以不同的方法考察仿制药与原研药的溶出度一致性,能更全面地评价国产仿制药与原研药溶出行为的差异。
猜你喜欢
中国医院用药评价与分析(2022年5期)2022-06-23
家庭医药·快乐养生(2022年6期)2022-06-23
中国外汇(2019年13期)2019-10-10
中国药房(2019年21期)2019-09-10
中国外汇(2019年7期)2019-07-13
大众健康(2019年6期)2019-06-10
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
智富时代(2018年6期)2018-08-06
智富时代(2018年6期)2018-08-06
