
缺氧预处理的脂肪间充质干细胞对小鼠硬皮病模型的治疗作用
2020-09-01 13:49李宇飞金佳慧沈亮亮BhavanaRajbanshi赵敬军
同济大学学报(医学版) 2020年4期
吴 晓,李宇飞,金佳慧,沈亮亮,Bhavana Rajbanshi,赵敬军
(1.同济大学医学院,上海 200092;2.同济大学附属同济医院皮肤科,上海 200065)
硬皮病是一种以皮肤和内脏器官纤维化为主要改变的慢性自身免疫性疾病,其进展的过程主要为小血管受损,导致免疫细胞浸润,进而发展为结缔组织功能异常,大量胶原沉积于病变区[1]。目前硬皮病的发病机制有多种学说[2],如1993年Murrell首次提出硬皮病可能与组织内氧化应激水平改变有关,后续大量研究也支持这一猜想[3]。脂肪间充质干细胞(adipose tissue-derived stromal cells, ADSCs)在硬皮病小鼠模型中被证明能够逆转硬皮病的进展,其机制研究主要集中在对皮损区的免疫调节以及旁分泌抗纤维化的细胞因子[4-5],而对于调节氧化应激水平研究较少。在有关干细胞移植的治疗研究中,有实验结果证实干细胞经缺氧预处理后能够增加其对疾病的治疗效果[6]。故本文对ADSCs和缺氧预处理的ADSCs(hypoxia-pretreated ADSCs, hpADSCs)治疗硬皮病小鼠的皮肤纤维化的逆转效果和对氧化应激水平的影响进行了初步研究。
1 材料与方法
1.1 动物和材料
SPF级4~6周龄雌性C57BL/6小鼠购自上海斯莱克实验动物有限责任公司,饲养于同济大学沪北实验动物中心;博来霉素(bleomycin, BLM)购自美国Sigma公司;Western印迹法相关试剂购自碧云天生物技术公司;兔抗鼠TGF-β多克隆抗体、兔抗鼠α-SMA多克隆抗体购自美国Abcam公司;H-E染色试剂盒、Masson染色试剂盒购自北京索莱宝科技有限公司;羟脯氨酸试剂盒、H2O2试剂盒购自南京建成生物工程研究所;胶原酶Ⅰ、DMEM培养基、胎牛血清、100U/mL青链霉素均购自美国Thermo Fisher公司;CD34、CD44、CD73、CD90、CD105流式抗体均购自美国BD公司。
1.2 硬皮病小鼠模型的构建
将实验小鼠按随机表法分为4组,分别为空白对照组、BLM模型组、ADSCs治疗组、hpADSCs治疗组,每组6只。处理前剃除小鼠背部毛发,空白对照组每只小鼠注射100μL PBS,其余3组每只小鼠皮下注射100μL博来霉素PBS溶液(1mg/mL),每日1次,连续28d。
1.3 ADSCs的分离鉴定培养
将小鼠断颈处死,在无菌条件下取双侧腹股沟脂肪组织并置于0.1%的 Ⅰ 型胶原酶溶液中。在37℃,120r/min摇床消化1h后离心(离心半径18cm,1000r/min,5min),弃上清液,加入含15%胎牛血清的DMEM培养基并接种于培养皿中,48h后更换培养基。待贴壁细胞生长至70%~80%融合度时进行传代培养。取第3代ADSCs在显微镜下拍照,流式细胞术鉴定细胞表面标志物CD34、CD44、CD73、CD90、CD105。
1.4 预缺氧处理ADSCs和ADSCs/hpADSCs移植实验
取第4代ADSCs,一半置于37℃含1%O2、5%CO2、94%N2的细胞培养箱中进行缺氧预处理2d,同时另一半细胞置于37℃含5%CO2、21%O2、74%N2的细胞培养箱中进行培养。培养2d后消化重悬2种细胞,并调整细胞密度均为2×107个/mL。ADSCs治疗组及hpADSCs治疗组小鼠每只分别注射100μL ADSCs或hpADSCs悬液。空白对照组和BLM模型组同时注射等体积的PBS溶液。
1.5 皮损区羟脯氨酸和H2O2检测
取小鼠部分皮损组织,依据试剂盒说明书的要求,对皮损区羟脯氨酸、H2O2含量进行检测。
1.6 Western印迹法
取小鼠皮损组织并加入RIPA裂解液,使用组织研磨仪研磨使其充分裂解后提取蛋白液。使用BCA法测定蛋白液中的总蛋白浓度。加入适当体积的5×蛋白上样缓冲液后在金属干浴仪中加热100℃ 10min。每孔上样20μg总蛋白,通过SDS-PAGE、转膜和封闭后,加入适当体积的TGF-β(1∶1000)和α-SMA(1∶500)抗体过夜。孵育结束后加入相应的二抗(1∶5000)室温下反应1h,随后进行ECL化学发光反应。
1.7 H-E、Masson及免疫组化染色
小鼠皮肤组织经4%多聚甲醛固定,进行常规脱水、石蜡包埋、切片。切片用二甲苯脱蜡,梯度乙醇水化,TBST(含0.1% Tween20的Tris-HCl缓冲盐溶液)冲洗3次。用于H-E、Masson染色的切片根据试剂盒说明书的要求进行染色。用于进行免疫组化染色的切片放入微波炉进行热修复,自然冷却至室温后用3% H2O2孵育10min,5%山羊血清封闭10min,用TBST冲洗3次后分别滴加兔抗鼠TGF-β单克隆抗体(1∶50)、兔抗鼠α-SMA单克隆抗体(1∶100),4℃过夜。次日TBST冲洗3遍后,分别加入辣根过氧化酶标记的山羊抗兔二抗(1∶500)在室温下孵育2h。TBST冲洗3次后用二氨基联苯胺显色,苏木精复染,中性树胶封片。胞质内出现棕黄色颗粒为阳性反应。
1.8 数据统计

2 结 果
2.1 ADSCs的形态观察与鉴定
ADSCs接种2d后开始明显的贴壁生长,第3代ADSCs细胞呈梭形或成纤维细胞状,细胞核清晰可见,见图1A。缺氧培养2d后,hpADSCs形态无明显改变,见图1B。流式鉴定ADSCs特征性表面分子:CD34(-)、CD44(+)、CD73(+)、CD90(+)、CD105(+),见图2。
图1 ADSCs、hpADSCs形态学特征(×40)

图2 ADSCs表面分子表达情况
2.2 BLM诱导的小鼠硬皮病模型的病理改变及ADSCs/hpADSCs治疗后变化
如图3所示,H-E染色可见空白对照组真皮层胶原纤维分布结构正常,脂肪层明显。而BLM模型组真皮层胶原明显增多,脂肪层消失,并伴有明显的炎性细胞浸润。ADSCs移植治疗后真皮层厚度无明显变化,但相对BLM模型组排列疏松,脂肪层明显增厚。hpADSCs治疗组的真皮层胶原增生情况明显改善,脂肪层明显增厚。Masson染色可见空白对照组皮肤结构正常,胶原排列有序。BLM模型组真皮明显增厚,胶原增生聚集且排列紊乱,见 图4。经ADSCs和hpADSCs治疗后,真皮变薄,胶原减少,与空白对照组类似。图5表明未处理的ADSCs治疗虽然在一定程度上降低了真皮厚度,但是与BLM模型组相比,差异没有统计学意义(P>0.05)。hpADSCs能够更好地降低皮损部位的胶原厚度,与BLM模型组相比,差异具有统计学意义(P<0.01)。
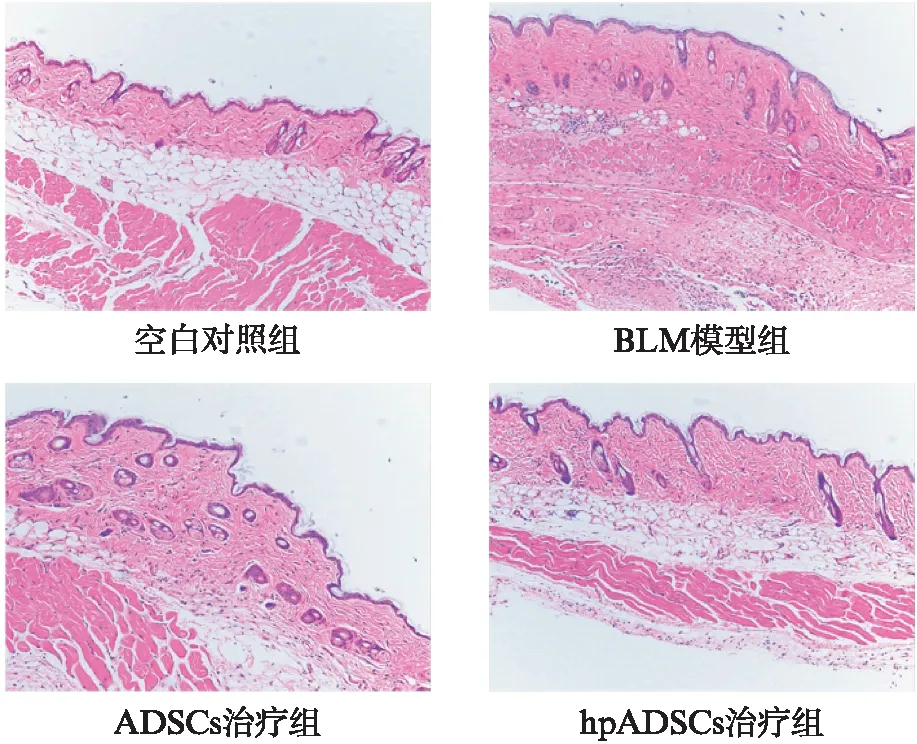
图3 皮损区样本H-E染色(×100)

图4 皮损区样本Masson染色(×100)
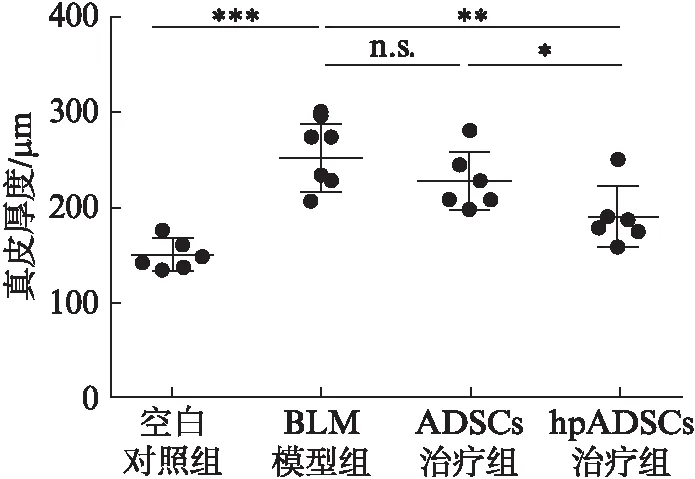
图5 各组样本皮损区真皮厚度
2.3 ADSCs/hpADSCs对硬皮病的胶原合成水平的影响
羟脯氨酸是胶原的主要成分之一且为胶原特有的氨基酸,其他蛋白中几乎不含有羟脯氨酸,故可以通过羟脯氨酸的量来间接评估胶原的含量。经BLM处理后皮肤组织内的羟脯氨酸表达水平明显变高,经ADSCs/hpADSCs治疗后皮损部位的羟脯氨酸水平均明显下调,与BLM模型组相比,差异有统计学意义(P<0.05)。hpADSCs治疗后样本的羟脯氨酸水平略低于ADSCs治疗组,但是差异无统计学意义(P>0.05),见图6。

图6 ADSCs及hpADSCs治疗对皮损部位胶原的影响
2.4 ADSCs/hpADSCs对皮损部位氧化应激水平的影响
组织内H2O2浓度能够反映组织内的氧化应激水平。检测了4组样本中的H2O2水平,发现BLM能够明显上调组织内的H2O2水平(图7),这与本团队之前发现的现象一致[7]。在干细胞治疗后皮肤内的H2O2减少,这提示了干细胞移植治疗能够下调硬皮病内氧化应激水平,并且与ADSCs相比,hpADSCs的抗氧化应激能力相对更佳。

图7 ADSCs及hpADSCs治疗对皮损部位H2O2水平的影响
2.5 hpADSCs对皮损部位TGF-β和α-SMA表达的影响
首先通过Western印迹法验证组织内TGF-β和α-SMA的水平。发现经过ADSCs/hpADSCs治疗后皮肤内TGF-β的水平均有一定的下降。α-SMA仅在hpADSCs组有一定的下调,见图8。为了进一步观察α-SMA和TGF-β在组织内的分布和表达情况,对小鼠石蜡切片进行了免疫组化染色。α-SMA表达于胞质中,阳性细胞散在分布于真皮层。BLM处理的小鼠真皮层α-SMA阳性细胞数明显增多。经ADSCs治疗后的小鼠真皮内α-SMA阳性细胞数变化不大,而hpADSCs组的α-SMA阳性细胞数明显减少,见图9。与空白对照组相比,BLM处理后的小鼠真皮层内TGF-β大量表达,且毛囊处的TGF-β增高显著。ADSCs治疗后真皮层中毛囊组织中TGF-β轻度下调,但是整体变化不大。hpADSCs治疗后显著降低皮肤中的TGF-β的水平,见图10。
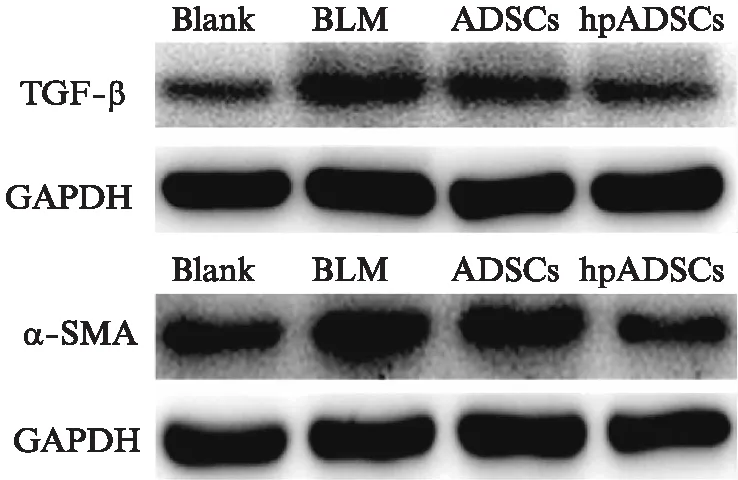
图8 Western印迹法分析TGF-β和α-SMA在小鼠皮肤中的表达情况

图9 皮损组织样本中α-SMA的表达情况(×100)
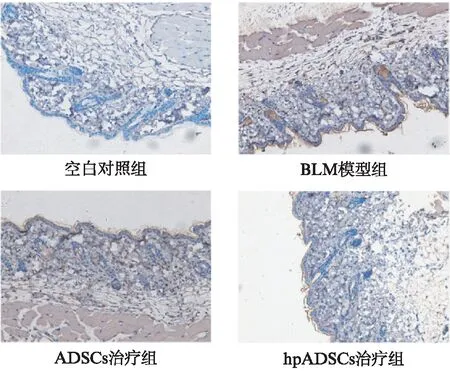
图10 皮损组织样本中TGF-β的表达情况(×100)
3 讨 论
硬皮病是一种以血管病变,免疫异常激活及胶原异常沉积为特点的自身免疫性疾病。血管内皮损伤在硬皮病的进展中是最早发生的[8]。血管内皮具有复杂的生物学功能,如组织血小板和白细胞的黏附,控制血管收缩扩张,抑制细胞外基质的沉积以及控制炎症反应。内皮损伤后会导致局部生理状态异常,其中异常之一就是活性氧的产生[3]。活性氧类(reactive oxygen species, ROS)包括超氧阴离子(O2·)、过氧化氢(H2O2)、羟基自由基(OH·)、一氧化氮(·NO)、过氧亚硝基(ONOO-)和次氯酸(HOCl)。硬皮病患者体内ROS的水平与正常人群相比有一定程度的升高。ROS诱导硬皮病的机制主要包括诱导促炎和促纤维化的细胞因子如血小板源性生长因子、TGF-β表达,刺激成纤维细胞的增殖和激活,以及胶原合成,最终导致皮损区胶原异常沉积[9-10]。并且动物实验也验证了ROS能够诱导出与硬皮病类似的皮损特点[11]。因此,ROS在硬皮病的发生发展中具有重要作用。
BLM诱导的皮肤纤维化模型被广泛用于硬皮病动物实验研究。本研究中,小鼠经BLM皮下注射4周后检测发现注射区的胶原、H2O2、α-SMA、TGF-β明显升高,与硬皮病患者的皮肤特点一致,证明BLM诱导的小鼠硬皮病模型可以较好地模拟硬皮病患者的皮肤特点,并且可用于硬皮病氧化应激相关的研究。
目前硬皮病尚无安全有效的治疗方案。近来动物实验研究发现间充质干细胞(mesenchyma stem cells, MSCs)移植能够降低硬皮病的胶原沉积,延缓硬皮病的进展[12]。ADSCs是MSCs的一种,同样具有干细胞的生物学特性。目前研究发现ADSCs对硬皮病的治疗机制主要包括抑制成纤维细胞胶原分泌,抑制皮损区免疫反应及增加血管内皮生长因子水平[13-16]。动物实验表明ADSCs能够降低心、肝、肺、肾等多种器官的纤维化水平,降低局部炎症水平,抑制胶原沉积[4]。氧化应激是硬皮病发病的重要机制之一,本研究中证实了ADSCs对硬皮病小鼠皮肤组织氧化应激水平的影响,发现注射了ADSCs后皮肤内H2O2水平明显下降,提示ADSCs能够通过降低氧化应激水平,在其他研究中同样报道了包括ADSCs在内的干细胞能够降低组织的氧化应激水平[17-18]。经ADSCs治疗后真皮层略变薄,皮下脂肪层变厚,胶原水平降低,有效地下调小鼠皮肤局部胶原水平,逆转皮肤病变。
据文献报道,在硬皮病患者中TGF-β和α-SMA均显著上调[19-20],TGF-β能够诱导成纤维细胞激活并促进其胶原合成[21-23]。α-SMA是肌成纤维细胞表达的蛋白,是成纤维细胞激活的标志。本实验中发现了BLM模型组中α-SMA阳性细胞数明显高于空白对照组,提示博来霉素诱导的硬皮病可能能够通过TGF-β介导成纤维细胞激活,促进胶原合成。ADSCs和hpADSCs移植治疗能够有效的下调BLM诱导的硬皮病皮损中的α-SMA、TGF-β的表达,提示这两种干细胞确实能够通过干预成纤维细胞的功能来逆转皮肤纤维化。
本研究发现ADSCs缺氧预处理后可以增强其对硬皮病的治疗作用,但其具体机制目前尚不明确。任何细胞的生理特性都与细胞类型、成熟度和环境有关。根据文献报道,在高灌注的器官内如肝、心、肾等,氧浓度约4%~14%,而在脂肪这一类缺氧组织中氧浓度甚至不到3%,提示了ADSCs在自然状态下是处于相对缺氧状态,而在细胞体外培养中ADSCs是处在富氧状态(约21%),这会对ADSCs的干细胞特性造成一定的影响。在缺氧环境下培养ADSCs可能增强其扩增、黏附和细胞因子的合成分泌能力,这可能是hpADSCs治疗效果更好的原因[24]。有研究分析了缺氧预处理后ADSCs细胞内的基因表达的改变,发现上调的基因主要功能富集在基质降解、下调细胞蛋白代谢途径以及调节蛋白水解,下调基因主要功能富集在炎症诱导和免疫趋化功能[25]。该文献数据显示IL-6在缺氧预处理后表达升高5倍。有文献报道IL-6能够诱导STAT3的激活,进而活化核因子E2相关因子2并促进其依赖的抗氧化应激因子的表达,下调ROS及超氧化物的水平[26]。这可能是hpADSCs具有更好治疗效果的机制之一。Xu等[27]通过筛查缺氧前后的ADSC外分泌细胞因子的含量变化发现,IL-10在缺氧处理后的ADSCs上清中含量明显升高,而IL-10能够通过恢复细胞抗氧化酶的活性来降低细胞氧化应激水平[28]。总之,缺氧诱导后的ADSCs能够改变ADSCs的基因表达谱,提升ADSCs移植后的抗氧化应激能力。相比对其他的基因编辑方法来提升ADSCs的功能,缺氧预处理ADSCs相对更加容易,对临床ADSCs移植治疗硬皮病等相关疾病有一定的指导意义。
猜你喜欢
中国中西医结合皮肤性病学杂志(2022年4期)2022-09-18
中国中西医结合皮肤性病学杂志(2022年2期)2022-05-11
皮革科学与工程(2022年1期)2022-01-15
婚育与健康(2021年14期)2021-10-18
皮肤病与性病(2021年3期)2021-07-30
中国美容医学(2021年1期)2021-03-12
健康博览(2020年4期)2020-06-08
大自然探索(2018年2期)2018-06-12
家庭医药(2018年2期)2018-02-09
食品工业科技(2014年13期)2014-03-11
