
高速逆流色谱分离纯化桑葚花色苷及其抗氧化活性
2020-08-22 08:06薛宏坤李鹏程刘成海
食品科学 2020年15期
薛宏坤,李鹏程,钟 雪,刘成海,李 倩
(1.东北农业大学工程学院,黑龙江 哈尔滨 150030;2.清华大学工程物理系,粒子与辐射成像教育部重点实验室,北京 100084;3.吉林农业大学食品科学与工程学院,吉林 长春 130112;4.大连大学生命科学与技术学院,辽宁 大连 116622)
桑葚为桑科桑属(Morus albaL.)的聚花果,果实柔软多汁、色泽诱人、风味独特,且含有丰富的活性蛋白、白藜芦醇、超氧化物歧化酶、花青素等活性成分[1],具有较高的医学和药用价值,备受人们喜爱。花青素是桑葚中最重要的活性物质,属于水溶性的天然色素,安全无毒,在植物细胞液中多数以糖苷键形成花色苷而存在,其基本结构单元是C6-C3-C6骨架构成的2-苯基苯并吡喃[2]。因花色苷独特的结构使其具有抗氧化、抗炎、抗肿瘤、预防心血管疾病和增强视力等功效[3]。近年来,植物花色苷所呈现的生物活性和药理作用引起广泛关注,特别是随着合成色素暴露引发越来越多的安全问题,天然色素资源的探寻和开发已逐渐成为国内外学者研究的热点。目前,虽然对花色苷生理活性研究较多,但已有的研究结果仍存在不确定性,因为大部分研究主要针对花色苷粗提物,而从单一花色苷组分获得的研究数据有限。因此为进一步明确花色苷单体的生理活性,亟需寻找能获得单一高纯度花色苷组分的分离纯化方法。
桑葚花色苷的有效富集和纯化是获得单一高纯度花色苷单体的关键步骤。桑葚花色苷粗提物中含有大量糖类、蛋白质类、脂类及色素等杂质,严重影响花色苷的生物学活性和纯度[4],为获得单一高纯度花色苷单体,必须要对花色苷粗提物进行分离和纯化。传统分离花色苷的方法主要用大孔树脂层析、凝胶层析、C18反向色谱等方法[5]。但以上纯化花色苷的方法均存在制备量小、重现性差等缺点,无法满足对大量高纯度花色苷的需求。
高速逆流色谱(high-speed counter-current chromatography,HSCCC)技术是利用物质在两相中分配系数的不同而实现分离的一种色谱技术,具有简单快捷、制备量大、分离效率高、重现性好、无不可逆吸附、成本低和样品回收完全等优点。因此,该技术被广泛应用于越橘[6]、紫甘薯[7]、木瓜花瓣[8]、玫瑰茄[9]、杨梅[10]等植物花色苷分离纯化。而采用HSCCC分离纯化桑葚花色苷及对其组分进行鉴定分析的研究报道有限。鉴于此,本研究在大孔树脂分离纯化桑葚花色苷的基础上,通过HSCCC技术进一步对其进行分离纯化,结合紫外-可见光谱、高效液相色谱-质谱联用(high performance liquid chromatography-mass spectrometry,HPLC-MS)技术及核磁共振(nuclear magnetic resonance,NMR)技术对分离纯化得到的组分进行鉴定,同时测定桑葚花色苷粗提取物和分离得到花色苷组分的体外抗氧化活性,以期为功能性食品和保健食品的开发提供理论依据。
1 材料与方法
1.1 材料与试剂
桑葚采摘于黑龙江大兴安岭地区,采摘时间2018年8~9月,经除杂、洗净、晾干后,于-18 ℃冰箱中冷冻保存。
乙醇、甲醇、甲酸(分析纯)、硫代巴比妥酸(thiobarbituric acid,TBA)、大豆卵磷脂(lecithin soybean,LS)、三氯乙酸(trichloroacetic acid,TCA)(分析纯) 国药集团化学试剂有限公司;乙腈(acetonitrile,ACN)、正丁醇(BuOH,BH)、甲基叔丁基醚(methyl tert-butyl ether,MTBE)、三氟乙酸(trifluoroacetic acid,TFA)(色谱纯) 天津市科密欧化学试剂有限公司;AB-8大孔树脂 天津市大钧科技有限公司。
1.2 仪器与设备
1100 HPLC、1290 HPLC-G6500四极杆飞行时间MS联用仪 美国Agilent公司;HSCCC-300C HSCCC仪上海同田生物技术股份有限公司;SHZ-D(III)型循环水式真空泵 巩义仪器有限责任公司;RE-52AA型旋转蒸发仪 上海亚荣生化仪器厂;XY-S650CT台式恒温超声波提取机 上海析宇仪器有限公司;Advance III NMR光谱仪 瑞士Bruker公司;UV-mini1240紫外-可见分光光度计 日本岛津公司。
1.3 方法
1.3.1 实验流程
实验设计流程为:

1.3.2 桑葚花色苷粗提物的制备
在预实验的基础上,采用体积分数60%乙醇溶液(含体积分数0.05%醋酸)作为提取溶剂。称取1.5 kg冷冻状态桑葚果于打浆机中进行打浆,然后称取1 kg果浆,按照料液比1∶4加入提取溶剂,调节pH值至2.0,同时设定超声功率300 W、提取温度50 ℃、提取时间25 min,提取结束后,过滤,将滤渣在同样条件下提取2 次,合并3 次所得滤液,35 ℃下旋转蒸发仪减压浓缩,回收乙醇;将浓缩液以1∶1的体积比再加入无水乙酸乙酯萃取3 次,收集水相,35 ℃下旋转蒸发仪减压浓缩,得到花色苷粗提液。花色苷粗提液以2 BV/h流速通过已活化的AB-8大孔树脂柱(2.6 cm×60 cm),待上样完成后,将其静置15 min,然后分别以2 BV的蒸馏水和3 BV体积分数40%、60%、80%酸化乙醇(含体积分数0.05%醋酸)依次洗脱,流速3 BV/h,收集体积分数60%乙醇洗脱液,将其用旋转蒸发仪于35 ℃减压浓缩、冷冻干燥,得到花色苷粗提物粉末61.5 g,避光密封保存在-18 ℃冰箱中。
1.3.3 HSCCC溶剂系统选择
参考Qiu Fang等[11]的方法选取BH-MTBE-ACN-水-TFA作为HSCCC溶剂体系,将不同溶剂按照比例进行配制,振摇后静置分层,上相为固定相、下相为流动相。根据化合物在两相中的分配系数不同,筛选最适的溶剂比例。准确称取0.1 g经1.3.2节制备的花色苷粗提物粉末溶解于上相(10 mL)与下相(10 mL)中,摇匀,待两相分配达到平衡后,将两相进行分离,分别各取1 mL,然后将其用0.45 µm膜过滤,用HPLC检测分析,依据式(1)计算不同溶剂的分配系数K。

式中:K为化合物在两相中的分配系数;A1为上相中化合物的峰面积;A2为下相中化合物峰面积。
1.3.4 HSCCC分离制备
按照1.3.3节筛选的最佳BH-MTBE-ACN-水-TFA比例组合为HSCCC溶剂体系,将配好的溶剂体系静置过夜后两相分离,超声脱气30 min。进样前,首先开启循环水浴仪,设定温度25 ℃、固定相流速15 mL/min,待固定相充满螺线管后,打开主机电源,缓慢正向调节转速至850 r/min,当转速恒定后,以2 mL/min流速泵入流动相,待整个体系建立完成后,准确称取花色苷粗提物粉末0.2 g用20 mL下相充分溶解,进样开始分离,检测波长254 nm,根据色谱流出图对组分出峰部位进行分步收集,流速3 mL/min,每管收集6 mL,对收集得到的组分进行冷冻干燥,且避光保存在-18 ℃冰箱中备用。
1.3.5 纯度测定与组分鉴定
1.3.5.1 紫外-可见光谱分析
采用质量分数0.03%氯化钾溶液分别溶解1.0 mg经1.3.4节HSCCC分离所得组分,然后分别将其在200~700 nm波长处进行光谱扫描[12],分析其光谱特征。
1.3.5.2 HPLC分析检测
样品处理:分别准确称取1.0 mg经1.3.4节HSCCC分离所得组分和花色苷粗提物粉末,并用体积分数0.1%HCl-甲醇溶液稀释后,配制成质量浓度为0.1 mg/mL的样品溶液,溶液过0.45 μm滤膜,用于HPLC分析。
色谱条件:1100 C18柱(150 mm×4.6 mm,2.6 μm);流动相A:甲酸-水(体积比1∶5);流动相B:乙腈-水-甲酸(体积比62∶40∶3);洗脱程序:10% B(0 min)~30% B(10 min)~30% B(15 min)~100% B(25 min)~100% B(30 min)~10% B(45 min);流速0.5 mL/min;柱温25 ℃;进样量20 μL;检测波长520 nm。依据峰面积归一法计算所得组分的纯度(以峰面积比例表示),计算公式如式(2)所示。

式中:wi为待测组分i的峰面积比例/%;fi为校正因子;Ai为待测组分i的峰面积。
1.3.5.3 HPLC-MS分析
HPLC条件:1290 C18柱(150 mm×2.1 mm,5 μm),流动相A:体积分数0.1%甲酸水溶液;流动相B:体积分数0.1%甲酸乙腈溶液;洗脱程序:15% B(0 min)~40% B(15 min)~60% B(20 min)~75% B(30 min)~90%B(35 min)~15% B(45 min);流速0.2 mL/min;柱温35 ℃;进样量20 μL;检测波长525 nm。
MS条件:电喷雾电离离子源,MS采用正离子扫描方式,质量扫描范围m/z100~1 000,毛细管电压4.5 kV,雾化器压力138 kPa,干燥气温度335 ℃。
1.3.5.4 NMR结构鉴定
样品处理:分别准确称取1.0 mg经1.3.4节HSCCC分离所得组分,并用氚代甲醇溶解,配制成质量浓度为0.1 mg/mL的样品溶液,溶液过0.45 μm滤膜,用于NMR分析。通过MS、1H-NMR和13C-NMR光谱数据库及参考文献,确定组分结构。
1.3.6 抗氧化能力测定
1.3.6.1 抗脂质体过氧化能力测定
参照樊金玲等[13]的方法并略作改动。分别准确称取2.0 mg样品(花色苷粗提物粉末、组分I、II、III),用体积分数60%酸化乙醇(含体积分数0.05%醋酸)将其配制成不同质量浓度(0.25、0.50、0.75、1.00、1.25 g/L和1.50 g/L)样品溶液。分别取1 mL样品溶液,分别加入1 mL LS溶液和1 mL 0.4 mol/L FeSO4溶液,充分混合。避光37 ℃恒温水浴1 h,再加入2 mL TCA-TBA-HCl(15 g TCA、0.37 g TBA、2.1 mL HCl)混合液,避光水浴15 min,然后以3 000 r/min离心10 min,取上清液在532 nm波长处测定其吸光度。以相同质量浓度VC溶液为阳性对照,依据公式(3)计算脂质体抑制率。

式中:A样品为样品组的吸光度;A对照为以1 mL VC溶液代替1 mL样品溶液,操作同上,测得的对照管吸光度。
1.3.6.2 1,1-二苯基-2-三硝基苯肼自由基清除能力测定
参照封燕等[14]的方法并略作改动。分别准确称取3.0 mg样品(花色苷粗提物粉末、组分I、II、III),采用体积分数60%酸化乙醇(含体积分数0.05%醋酸)将其配制成不同质量浓度(0.10、0.20、0.40、0.60、0.80 g/L和1.00 g/L)样品溶液。分别取出2 mL置于6 个10 mL具塞比色管中,分别加入2.8 mL 1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-trinitrophenylhydrazine,DPPH)溶液并将其充分混合,避光室温下放置30 min,在517 nm波长处分别测定其吸光度。以相同质量浓度VC溶液为阳性对照,由公式(4)计算DPPH自由基清除率。

1.4 数据统计分析处理
每组实验重复3 次,结果用平均值±标准差表示;采用Statistix 8.0软件对每组实验数据进行方差分析;采用SAS 8.0软件分析结果的差异显著性,以P<0.05表示差异显著;通过Origin 9.0软件绘图,Chemical Draw 15.0软件绘制花色苷结构。
2 结果与分析
2.1 HSCCC溶剂体系的确定
采用HSCCC技术分离天然产物最关键的步骤就是确定合适的溶剂体系,参考文献[15-16],本研究采用BH-MTBE-ACN-水-TFA为溶剂体系,并采用HPLC法快速、高效地测定K,确定最适的HSCCC溶剂体系配比,各溶剂配比及K如表1所示。

表1 不同溶剂体系配比的分配系数Table 1 Distribution coefficients (K) of different solvent systems
由表1可知,溶剂体系2、3、4各组分的K均在0.5~2.0范围内,符合HSCCC溶剂体系配比,但在2、3、4体系中,不同组分K无显著差异(P>0.05),导致4 种组分不能完全分开,故不适合作HSCCC溶剂体系。体系5中各组分的K较小,组分分离效果较差,因此,体系5不适合作为HSCCC溶剂体系。体系1各组分的K均在0.5~2.0,且各组分的K具有显著差异(P<0.05),组分分离效果较好,结果说明体系1适合作为HSCCC溶剂体系。该结果与Chen Liang[17]和Li Bo[18]等利用HSCCC技术分别从蓝果忍冬和黑米糠分离花色苷所用HSCCC溶剂体系配比一致。
2.2 HSCCC分离纯化结果
依据1.3.4节的方法,以BH-MTBE-ACN-水-TFA(2∶2∶1∶5∶0.01,V/V)作为HSCCC溶剂体系,桑葚花色苷经HSCCC分离所得实验结果如图1所示。

图1 HSCCC法分离桑葚花色苷的色谱图Fig.1 HSCCC chromatogram of anthocyanins separated from mulberry fruit
由图1可知,桑葚花色苷经HSCCC分离,一共得到4 个组分,分别命名为组分I~IV,同时测定固定相的保留率为52.6%,说明所用HSCCC溶剂体系可行,且目标成分分离良好。从200 mg花色苷粗提物粉末中分离得到组分I(34.8 mg)、组分II(67.4 mg)、组分III(19.6 mg)、组分IV(16.3 mg)。
2.3 紫外-可见光谱分析结果
经HSCCC分离所得的组分I~IV分别用质量分数0.03%氯化钾溶液溶解,并稀释至一定质量浓度,然后用紫外-可见光谱仪进行扫描,其吸收光谱如图2所示。
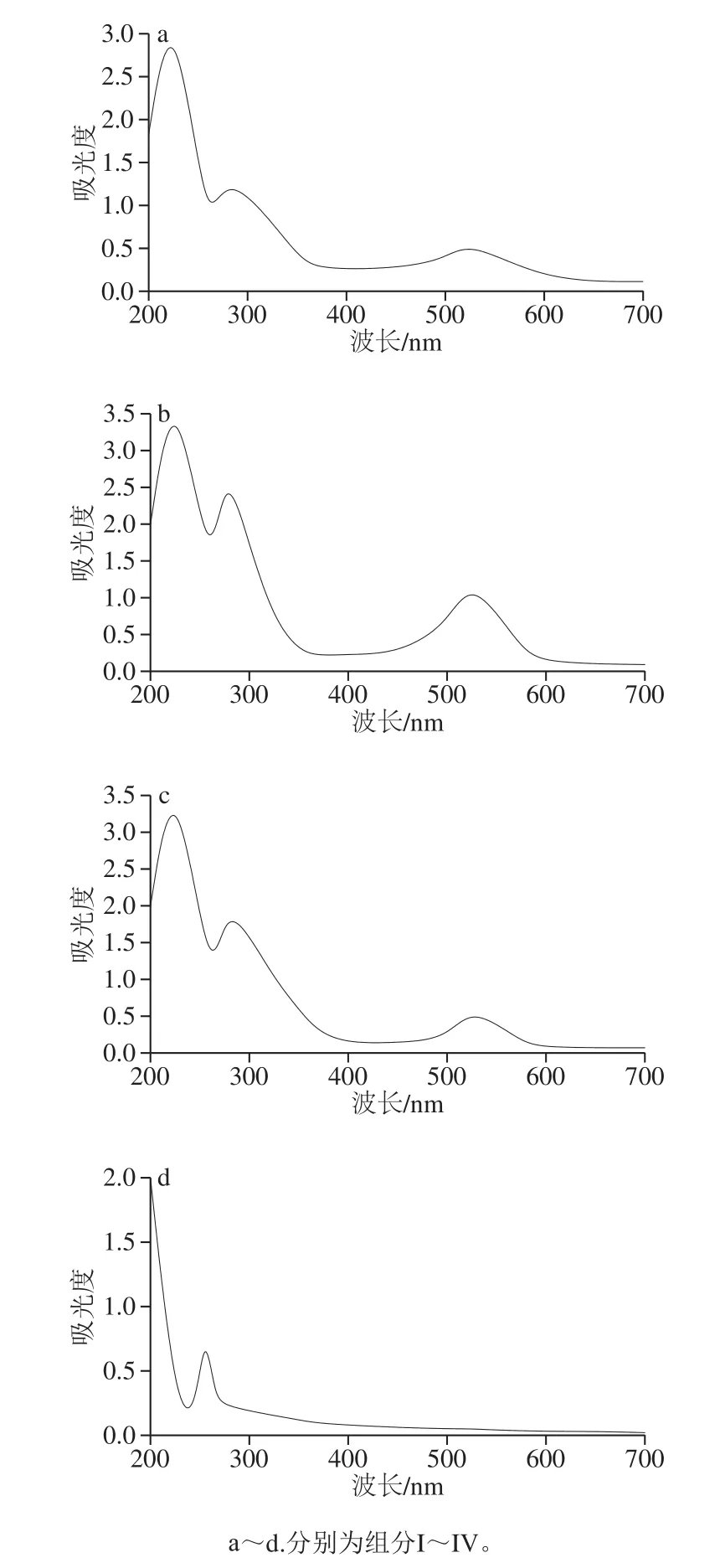
图2 HSCCC法分离桑葚花色苷组分的紫外-可见光谱Fig.2 UV-Vis spectra of anthocyanin components separated from mulberry fruit by HSCCC
花色苷属于多羟基的黄酮类化合物,分别在波长280 nm和520 nm左右具有特征吸收峰,而其他黄酮类化合物(黄酮醇、黄酮、黄烷酮等)在波长300~400 nm和240~280 nm的紫外区具有特征吸收[19]。因此,可以根据组分在波长280 nm和520 nm左右有无吸收特征,作为判断是否为花色苷类物质的依据。由图2a~c可知,组分I~III均在波长280 nm和520 nm左右具有特征吸收峰,说明组分I~III均为花色苷类物质。由图2d可知,组分IV在255 nm波长处具有特征吸收峰,且在波长280 nm和520 nm左右均无特征吸收峰,说明组分IV为非花色苷的黄酮类化合物,由于本研究的主要目的是分离纯化桑葚花色苷,故组分IV不再进行后续的研究。
2.4 HPLC检测分析结果
采用HPLC对桑葚花色苷粗提取粉末和经HSCCC分离所得的花色苷组分(组分I、II、III)进行纯度检测分析,其结果如图3所示。

图3 桑葚花色苷粗提物及组分I、II、III的HPLC图Fig.3 HPLC chromatograms of crude extract, and components I, II and III
由图3可知,桑葚花色苷粗提物包含3 种花色苷组分,且分离效果较好。依据出峰时间确定组分I~III分别对应图3a中的1~3号峰。另外,组分I~III的HPLC图均无杂峰,表明组分I~III均为单一的花色苷组分。根据峰面积计算组分I~III的纯度分别为92.27%、94.05%和90.82%。采用HPLC-MS和NMR技术进一步鉴定以上3 种花色苷组分。
2.5 HPLC-MS和NMR检测分析
为确定组分I~III的结构,利用HPLC-MS和NMR技术对其进行鉴定。根据一级MS、二级MS、1H NMR和13C NMR数据来确定花色苷的结构,上述3 种花色苷组分的MS图见图4,其鉴定结果如表2所示。
由表2可知,组分I保留时间为13.72 min,图4a1、a2中的MS和MS2信息表明,其分子离子峰[M+]为465.1,在MS中检测到碎片离子为m/z303.1。m/z303是飞燕草素的特征离子,丢失的碎片为m/z162,可能是失去葡萄糖和半乳糖;另外组分I的紫外-可见光谱显示其可见光谱最大波长为523 nm,且A440nm/Aλmax为28.0%,结合文献[20-21]分析,初步可判定组分I为飞燕草素-3-葡萄糖苷。将组分I进一步由NMR分析,1H NMR(CF3COODCD3OD):δ6.66(1H, d,J=1.2 Hz, H-6)、6.87(1H, d,J=1.2 Hz, H-8)、7.74(2H, d,J=2.2 Hz, H-2', 6')、8.93(1H, s, H-4);5.34(1H, d,J=7.8 Hz, H-1 glc)、3.73(1H, m, H-2 glc)、3.60(2H, m, H-3,5 glc)、3.51(1H, m, H-4 glc)、3.79(1H, dd,J=12.2, 5.4 Hz, H-6b glc)、3.94(1H, dd,J=12.2, 2.0 Hz, H-6a glc)。13C NMR(CF3COODCD3OD):δ164.2 (C-2)、144.6(C-3)、136.8(C-4)、159.4(C-5)、103.1(C-6)、170.8(C-7)、95.4(C-8)、157.8(C-9)、113.0(C-10)、121.5(C-1')、113.7(C-2')、147.6(C-3')、146.0(C-4')、147.9(C-5')、113.4(C-6')、103.5(C-1 glc)、74.8(C-2 glc)、78.0(C-3 glc)、71.2(C-4 glc)、78.8(C-5 glc)、62.3(C-6 glc)。组分I的1H NMR在δ6.66(2H, d)和δ6.87(2H, d)存在二重质子峰,耦合常数J=1.2 Hz,表明A环上存在两个超耦合的双重质子,推测为H-6和H-8。在δ7.74(2H, d,J=2.2 Hz)下观察到一组AM系统的双峰,这是H-2'和H-6'的特征。δ5.34(1H, d,J=7.8 Hz)表明组分I仅含有一个单葡萄糖,同时在δ144.6处与C-3配基结合。该研究结果与Alexandra等[22]研究香蕉苞花色苷飞燕草素-3-葡萄糖苷的NMR信息相一致,故最终确定组分I为飞燕草素-3-葡萄糖苷。


图4 花色苷组分质谱图Fig.4 MS of anthocyanin components
由表2可知,组分II保留时间为21.84 min,图4b1、b2中的MS和MS2信息表明,其分子离子峰[M+]为449.1,在MS中检测到碎片离子为m/z287.2。m/z287是矢车菊素的特征离子,丢失的碎片162可能是失去葡萄糖和半乳糖;另外组分II的紫外-可见光谱显示其可见光谱最大波长为516 nm,且A440nm/Aλmax为20.0%,参考文献[23-24],初步判定组分II为矢车菊素-3-葡萄糖苷。将组分II进一步用NMR分析,1H NMR(CF3COOD-CD3OD):δ6.64(1H, d,J=1.2 Hz, H-6)、6.86(1H, d,J=1.2 Hz, H-8)、8.22(1H, dd,J=8.8, 2.2 Hz, H-6')、8.02(1H, d,J=2.2 Hz, H-2')、7.04(1H, d,J=8.8 Hz, H-5')、8.98(1H, s, H-4)、5.31(1H, d,J=7.8 Hz, H-1 glc)、3.69(1H, m, H-2 glc)、3.56(2H, m, H-3,5 glc)、3.46(1H, m, H-4 glc)、3.73(1H, dd,J=12.2, 5.6 Hz, H-6b glc)、3.96(1H, dd,J=12.2, 2.2 Hz, H-6a glc)。13C NMR (CF3COODCD3OD):δ164.5(C-2)、145.8(C-3)、137.2(C-4)、159.6(C-5)、103.7(C-6)、170.8(C-7)、95.4(C-8)、157.9(C-9)、113.6(C-10)、121.5(C-1')、118.7(C-2')、147.6(C-3')、156.0(C-4')、117.7(C-5')、128.4(C-6')、104.1(C-1 glc)、75.0(C-2glc)、78.3(C-3glc)、71.3 (C-4 glc)、79.0(C-5 glc)、62.6(C-6 glc)。组分II的1H NMR表明,在δ6.64和δ6.86时,A环上存在两个超耦合的双重质子(J=1.2 Hz),分别分配给H-6和H-8。在δ7.04(1H, d,J=8.8 Hz)、8.22(1H, dd,J=8.8, 2.2 Hz)、8.02(1H, d,J=2.2 Hz)时,观察到两组双峰和一组ABM系统的双峰,分别具有H-5'、H-6'和H-2'的特征。环C是黄烷酮的一部分,且在δ3.46~5.31处的质子信号(H-1-glc-H-6-glc),表明组分II仅含有一个D-吡喃葡萄糖,在组分II的1H异核多碳相关谱中,在δ5.31/145.8(H-1 glc/C-3)处的交叉峰表明葡萄糖链接在C-3上[25]。Lee等[26]研究鹅掌楸果实花色苷的矢车菊素-3-葡萄糖苷的NMR信息与组分II一致,故最终确定组分II为矢车菊素-3-葡萄糖苷。
由表2可知,组分III保留时间为28.39 min,图4c1、c2中的MS和MS2信息表明,其分子离子峰[M+]为433.1,在MS中检测到碎片离子为m/z271.0,m/z271碎片离子是[M+H-162]所得,同时m/z271是天竺葵素的特征离子,且组分III的紫外-可见光谱显示其可见光谱最大波长为516 nm。参考文献[27],组分III初步判定为天竺葵素-3-葡萄糖苷。将组分III进一步由NMR分析,1H NMR(CF3COOD-CD3OD):δ6.67(1H, d,J=1.2 Hz, H-6)、6.94(1H, d,J=1.2 Hz, H-8)、7.05(2H, d,J=8.8 Hz, H-3', 5')、8.60(2H, d,J=8.8 Hz, H-2', 6')、9.08(1H, s, H-4)、5.34(1H, d,J=8.0 Hz, H-1 glc)、3.68(1H, m, H-2 glc)、3.54(2H, m, H-3,5 glc)、3.42(1H, m, H-4 glc)、3.80(1H, dd,J=12.2, 6.0 Hz, H-6b glc)、4.04(1H, dd,J=12.2, 2.0 Hz, H-6a glc)。13C NMR(CF3COODCD3OD):δ164.8(C-2)、145.8(C-3)、137.4(C-4)、159.9 (C-5)、103.7(C-6)、170.9(C-7)、95.5(C-8)、158.1(C-9)、114.5(C-10)、121.5(C-1')、136.8(C-2')、118.6(C-3')、166.0(C-4')、118.7(C-5')、136.4(C-6')、104.1(C-1 glc)、75.3(C-2 glc)、78.5(C-3 glc)、71.3(C-4 glc)、79.0(C-5 glc)、62.6(C-6 glc)。同理,按照上述方式对组分III结构进行解析,同时参考组分III的相关NMR文献[28],最终确定组分III为天竺葵素-3-葡萄糖苷。

表2 桑葚花色苷种类鉴定结果Table 2 Identification of anthocyanins from mulberry fruit
2.6 抗氧化分析结果
2.6.1 脂质体过氧化能力的结果分析
卵磷脂通常被作为细胞模型用于体外脂质体过氧化实验,卵磷脂内含有高密度脂蛋白、低密度脂蛋白和极低密度脂蛋白。在Fe3+的催化下,其低密度脂蛋白和极低密度脂蛋白中的不饱和脂肪酸能发生过氧化反应。因此,可以通过Fe3+诱导卵磷脂体过氧化模型来评价桑葚花色苷的抗氧化活性[29]。在卵磷脂过氧化实验中,在532 nm波长处检测TBA反应物质吸光度可以反映脂质体过氧化程度。桑葚花色苷粗提取物、组分I~III和VC对抑制脂质过氧化能力的测定结果如图5所示。
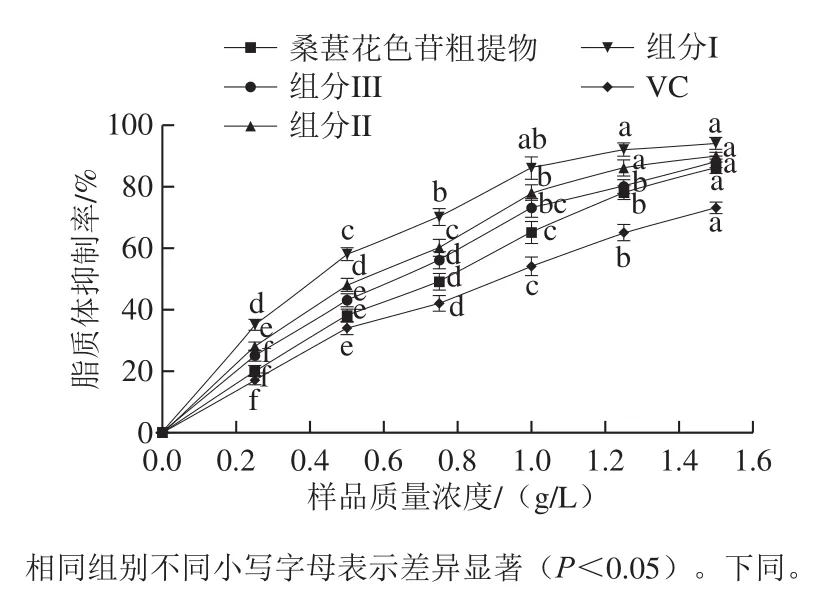
图5 桑葚花色苷和VC对脂质体抑制率的影响Fig.5 Inhibitory effect of mulberry anthocyanins and VC on lipid peroxidation
由图5可知,桑葚花色苷粗提物、组分I~III和VC均对脂质体过氧化有抑制作用,且随样品质量浓度的增加,脂质抑制率显著增加(P<0.05)。桑葚花色苷粗提物、组分I~III和VC抑制脂质体过氧化的半抑制浓度(50% inhibiting concentration,IC50)分别为(0.77±0.02)、(0.34±0.02)、(0.55±0.04)、(0.68±0.01)g/L和(0.91±0.03)g/L。分析样品的IC50可知,样品的抗氧化能力顺序为组分I>组分II>组分III>桑葚花色苷粗提物>VC。其可能原因是组分I结构上还原性酚羟基多于组分II和组分III。由于还原性酚羟基能够直接与酶和非酶系统产生的氧自由基发生反应,避免发生脂质体过氧化反应而产生脂质过氧化物;另外,由于还原性酚羟基产生共轭效应,氧原子上不成对的电子靠近苯环,使氢氧键作用力减弱,造成酚羟基氢原子活性提高,易于脱氢而形成供氢体,供氢体能与脂类化合物的自由基发生反应,转化成非常稳定的酚自由基,从而降低自动氧化链反应的传递速率,抑制脂类进一步氧化,故使抗脂质体过氧化能力增强[30]。组分I、II、III对脂质体抑制率高于桑葚花色苷粗提物。其原因是桑葚花色苷粗提物中含有不具有抗氧化能力的杂质,因此降低其对脂质体抑制率。
2.6.2 DPPH自由基清除能力分析结果
DPPH自由基是一种含有氮元素的稳定自由基,在醇溶液中呈现紫色,在517 nm波长处对紫外光有最大吸收[31],本研究通过DPPH自由基清除能力来判断花色苷的抗氧化能力。

图6 桑葚花色苷和VC对DPPH自由基清除率的影响Fig.6 DPPH radical scavenging effect of mulberry anthocyanins and VC
由图6可知,随组分I、II、III质量浓度增加,DPPH自由基清除率呈现先显著增加后无显著变化的趋势。桑葚花色苷粗提物、组分I、II、III和VC对DPPH自由基清除率的IC50分别为(0.40±0.01)、(0.16±0.01)、(0.22±0.01)、(0.35±0.03)g/L和(0.59±0.02)g/L。IC50越低表示抗氧化活性越强。对比IC50可知,组分I对DPPH自由基清除能力最强,且桑葚花色苷粗提物和组分I、II、III对DPPH自由基清除能力均高于VC。组分I对DPPH自由基清除能力优于组分II和组分III,其主要原因与其花色苷单体的结构相关,组分II和组分III苯环上还原性酚羟基的数目少于组分I,且组分III还原性酚羟基的数目少于组分II,在氧化过程中,还原性酚羟基作为主要的还原部位,能作为供氢体与氧化过程中产生的DPPH自由基发生反应,自身形成的自由基可以通过分子内氢键、半醌式自由基等形式得以稳定,从而中断自由基链的反应[32]。因此,花色苷组分对DPPH自由基清除能力大小顺序为组分I>组分II>组分III。
3 结 论
桑葚花色苷粗提物经HSCCC分离纯化后,得到4 种组分,并经紫外-可见光谱、HPLC-MS和NMR鉴定发现组分IV为非花色苷类,组分I~III分别为飞燕草素-3-葡萄糖苷、矢车菊素-3-葡萄糖苷和天竺葵素-3-葡萄糖苷,其含量和纯度分别为17.4、33.7、9.8 mg/100 mg和92.27%、94.05%、90.82%。桑葚花色苷对脂质体抑制和DPPH自由基清除能力大小顺序均为组分I>组分II>组分III>桑葚花色苷粗提物。本研究建立了从桑葚中分离纯化高纯度花色苷的HSCCC方法,同时为花色苷的进一步药理活性研究提供重要的物质基础。
猜你喜欢
中国食品学报(2022年8期)2022-09-07
江苏农业学报(2022年4期)2022-09-07
昆明医科大学学报(2022年3期)2022-04-19
中国果业信息(2021年5期)2021-12-05
现代临床医学(2021年6期)2021-11-20
昆明医科大学学报(2021年3期)2021-07-22
中国药学药品知识仓库(2021年18期)2021-02-28
食品安全导刊(2019年27期)2019-12-09
信息技术时代·上旬刊(2019年4期)2019-09-10
饮食科学(2016年12期)2017-01-07
