
饲料添加剂磷酸氢钙中铬的检测方法探究
2020-08-18 08:20何小华朱艳锦
饲料博览 2020年7期
李 芳,罗 雄,何小华,余 敏,朱艳锦
(成都蜀星饲料有限公司,成都 610000)
铬广泛存在于饲料原料及添加剂中,是动物必需的微量元素,机体中铬含量约为0.1 mg·kg-1,过量的铬会对机体造成危害。通常情况六价态的无机铬会对机体造成慢性中毒,严重影响畜禽生产,且在机体沉积后对食品安全造成威胁,因此GB13078-2017 中严格规定饲料原料及饲料产品中铬最高限量,以确保饲料产品安全[1]。GB22549-2017 中也规定磷酸氢钙中铬含量≤30 mg·kg-1[2]。笔者通过长期实验室检测发现,按照国家标准进行检测,不同的称样量,不同加热时长以及加热方式会导致其检测结果出现极大差异,结果不稳定。本文参考GB13078-2017、GB 22549-2017、NY/T 916-2004,对饲料添加剂磷酸氢钙中铬的测定部分条件进行探讨改进[3-4]。
1 材料与方法
1.1 方法
样品用酸溶解后,注入火焰原子吸收光谱检测器中检测。
1.2 仪器与试剂
电热炉、原子吸收分光光度计、盐酸(1+1)、硝酸(1+1)、硝酸(2+98)、10%氯化铵。
1.3 技术路线
1.3.1 标准曲线
标准曲线设置3组浓度梯度和2种溶液体系:1组、2 组标准液中含铬0、1.25、2.5、5、10、20 μg·mL-1;3 组、4 组标准液中含铬0、0.1、0.2、0.4、0.8、1.6 μg·mL-1,5 组、6 组标准液中含铬0、0.5、1.0、2.0、4.0、8.0 μg·mL-1,其中1组、4组、6组加入4 mL氯化铵(10%)。
试验设计见表1。

表1 标准曲线设计
1.3.2 样品前处理设计
设置两个试验组:国标组和改进组。国标组(GB 22549-2017)分为6个小组,具体操作:称10 g样品,加20 mL 盐酸(1+1),加热溶解,定容到250 mL,过滤为1号样液。取4个100 mL容量瓶分别标上A、B、C、D,分别移取1号液25 mL至容量瓶A 和B,5 mL 至C 和D,然后B 和D 中加入4 mL 10%氯化铵,定容;再取2 个50 mL 容量瓶标上E、F,各移取25 mL1 号液,F 加入2 mL10%氯化铵溶液,定容。改进组:G组称取1.5 g,H组称取3 g,加20 mL 硝酸(1+1),加盖表面皿,中火加热蒸至溶液剩余约8 mL,冷却转移,定容到50 mL,过滤。同时做样品空白。上机测定,同一个样品连续进样10次。试验设计见表2。

表2 称样量及前处理试验设计
2 结果分析
2.1 标准曲线结果
按照NY/T 916-2004 饲料添加剂吡啶甲酸铬和国家标准GB/T 13088-2006规定的相关条件进行检测,检测结果见图1。在实际检测过程中20 μg·mL-1检测结果偏差较大,影响标准曲线线性关系,可能原因是原子吸收分光度计极为灵敏,当浓度过大时吸光度与浓度偏离郎伯-比尔定律,也可能是浓度较大时铬离子原子化不完全。该结论与卢启明等研究一致,采用火焰原子吸收直接测定样品中铬含量,其铬含量在0~6 μg·mL-1时与吸光度线性相关性良好[5];此外,刘先华等研究指出铬元素在0.4~2.5 mg·L-1范围内线性关系良好[6]。由图1可知,NY/T 916-2004 的标准曲线较GB/T 13088-2006 吸光度显著提高,说明添加氯化铵可以提高吸光度。
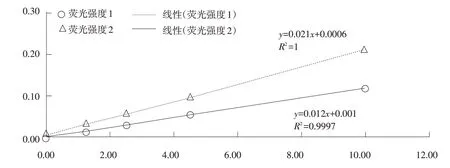
图1 1组和2组浓度和荧光强度的线性关系标准曲线
国家标准GB22549-2017 中规定铬的上限为30 mg·kg-1,称样量为2~10 g,根据操作步骤可知样品溶液的上限为0.3 μg mL-1,根据标准给出的火焰原子吸收法检出限(0.15 mg·kg-1),结合图1将标准曲线调整梯度进行调整,结果见图2。可见两组线性关系均较差。
经反复试验得出该方法定量限为0.5 μg·mL-1,进一步改进的结果见图3,试验5 组线性关系良好,回归方程为y=0.0104x+0.0003,R2=0.9992,符合检测要求。
2.2 称样量及前处改进结果与分析
检测结果见表3,由此可知国标组:A 组和B组存在差异,说明添加氯化铵影响铬的检测;B组检测结果G和H组结果相近,但B组的标准差分别是G组和H组1.9倍、5.4倍,说明硝酸有利于检测的稳定;此外,加氯化铵后样品加标回收高达127%,不符检测要求。C组和D组检测结果均不可靠,不符合检测要求。E、F 组其结果显著低于G 组和H组,说明称样量为10 g时样品溶解可能不够充分。
改进组:G组和H组检测结果相近,标准差小于1,样品加标回收率94%~108%,符合检测要求。由此可知,当样品浓度大于0.5 μg·mL-1时检测结果最为稳定,这与实验室自检定量检出限一致,与刘先华等研究结果也一致[6]。
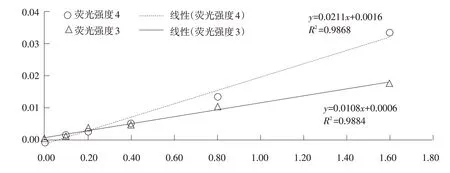
图2 3组和4组浓度和荧光强度的线性关系标准曲线

图3 5组和6组浓度和荧光强度的线性关系标准曲线
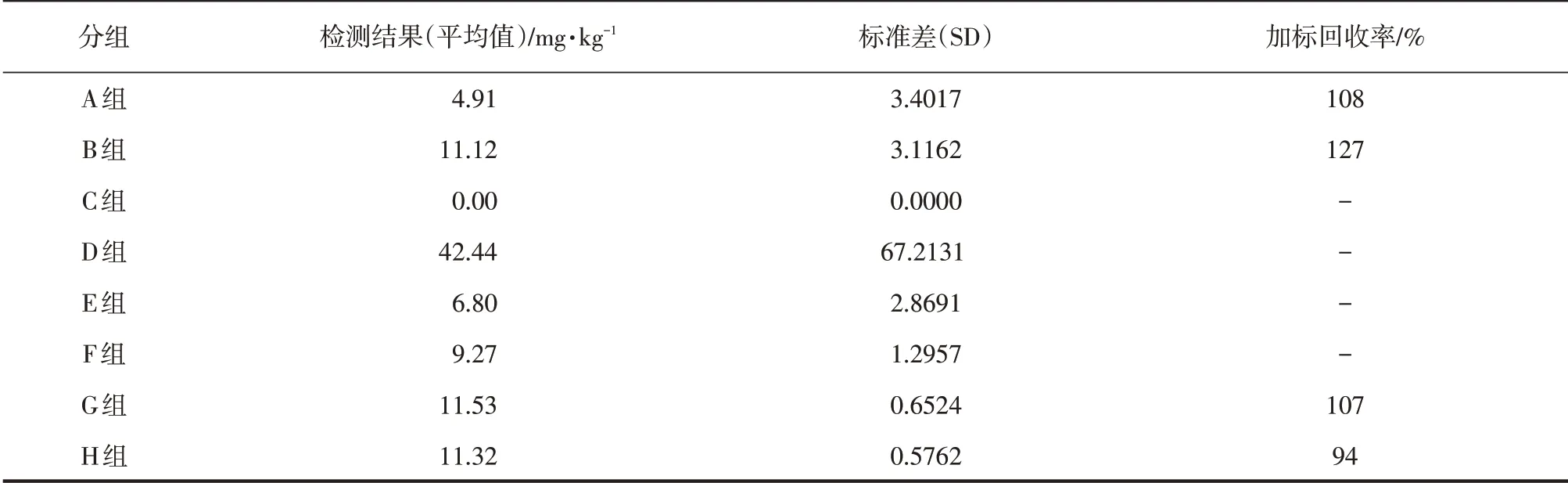
表3 铬含量值
3 结 论
试验结果表明,按照当前国家标准《饲料添加剂磷酸氢钙》(GB 22549-2017)处理步骤难以准确检测样品中铬含量,主因有:①提取不充分,计算可知10 g磷酸氢钙溶解理论上需要消耗近10 mL盐酸,难以保证样品充分溶解,与于家丰等研究结果一致[7];②实际生产中样液难以达到该方法定量检出限0.5 μg·mL-1,检测结果稳定性较差甚至无法检出。例如:当称样量为2 g 时,即便铬含量高达30 mg·kg-1,检测样液中铬浓度仅0.06 μg·mL-1,远低于标准的检出限0.15 μg·mL-1;③盐酸作为溶剂与标准体系不一致,且酸含量较硝酸低,溶解较差。
本研究通过改进样品前处理方法与步骤以及调整标准曲线浓度梯度,可充分溶解磷酸氢钙及样品中的铬,标准曲线线性关系更优,提高检测液含量浓度且达到方法要求,检测结果更加接近真实值,为改进完善国标方法提供数据参考。
猜你喜欢
出版人(2022年8期)2022-08-23
食品安全导刊(2020年33期)2020-12-04
食品安全导刊(2020年21期)2020-12-03
英语文摘(2020年6期)2020-09-21
无机盐工业(2019年12期)2019-12-12
食品安全导刊(2017年12期)2018-01-04
安徽农学通报(2017年20期)2017-11-10
Coco薇(2015年10期)2015-10-19
绿色科技(2015年4期)2015-07-14
农村农业农民·B版(2014年6期)2014-08-08
